EpiTect Hi-C Kit
适用于染色质折叠的高分辨率图谱绘制、基因组序列的高质量组装、单体型分型和染色体重排的鉴定
适用于染色质折叠的高分辨率图谱绘制、基因组序列的高质量组装、单体型分型和染色体重排的鉴定
✓ 全天候自动处理在线订单
✓ 博学专业的产品和技术支持
✓ 快速可靠的(再)订购
Cat. No. / ID: 59971
✓ 全天候自动处理在线订单
✓ 博学专业的产品和技术支持
✓ 快速可靠的(再)订购
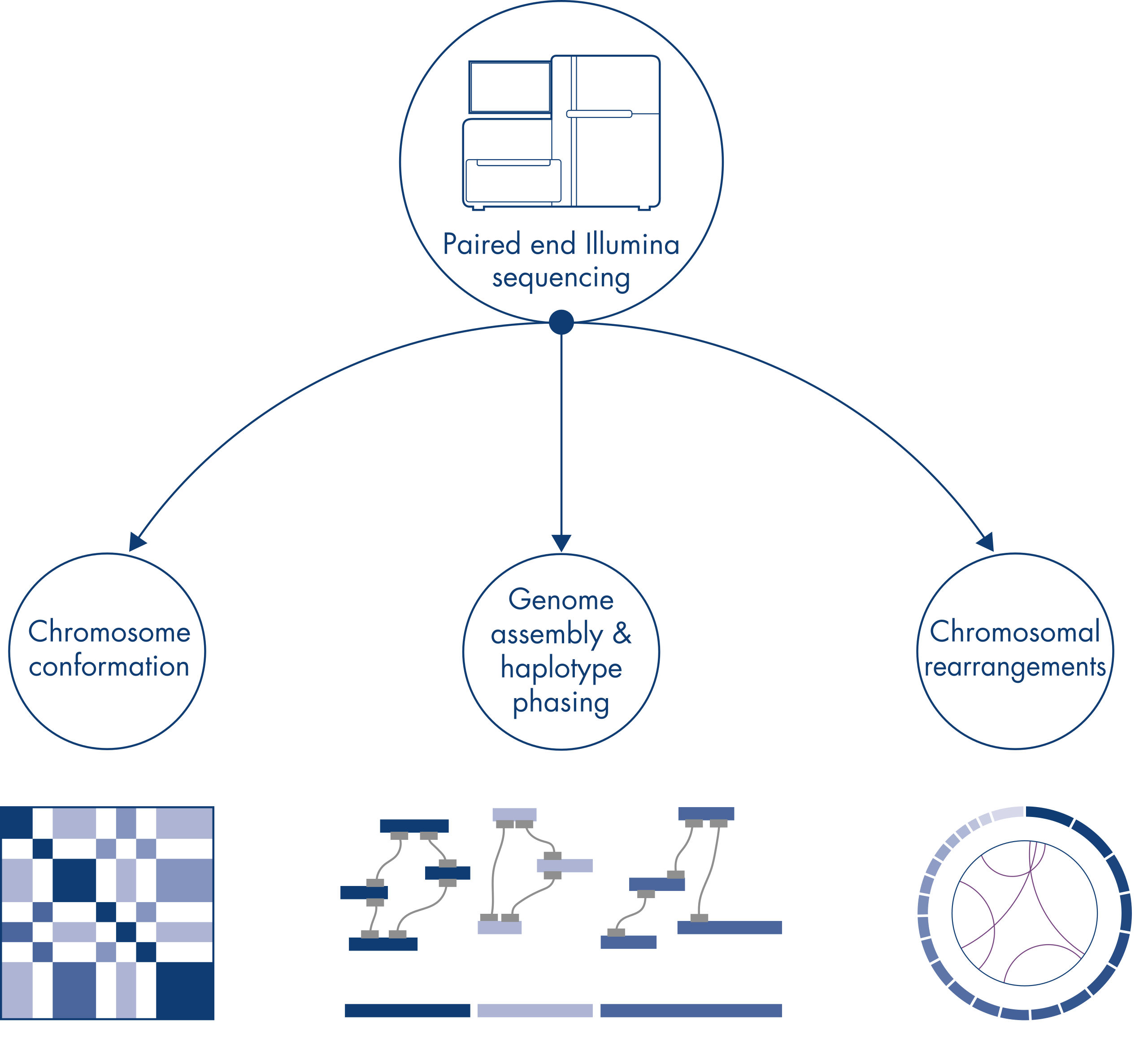
Hi-C 最初被视为全基因组染色体构象捕获的强大技术,能以 kb 分辨率鉴定染色质折叠。不过,该技术还有其他重要应用。例如,Hi-C 可用于从没有已知参考基因组的生物体中生成高度连续的基因组组装体,极少数组装体拥有很长的 scaffolds。此外,Hi-C 对单体型分型和染色体重排检测也非常有用。
EpiTect Hi-C Kit 提供了一个稳健而又简单快速的方案,细胞投入要求低,能在不到 2 天的时间内从交联细胞中生成高质量的 Hi-C Illumina NGS 文库。
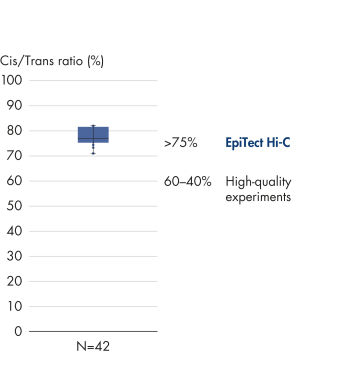
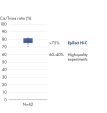
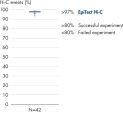
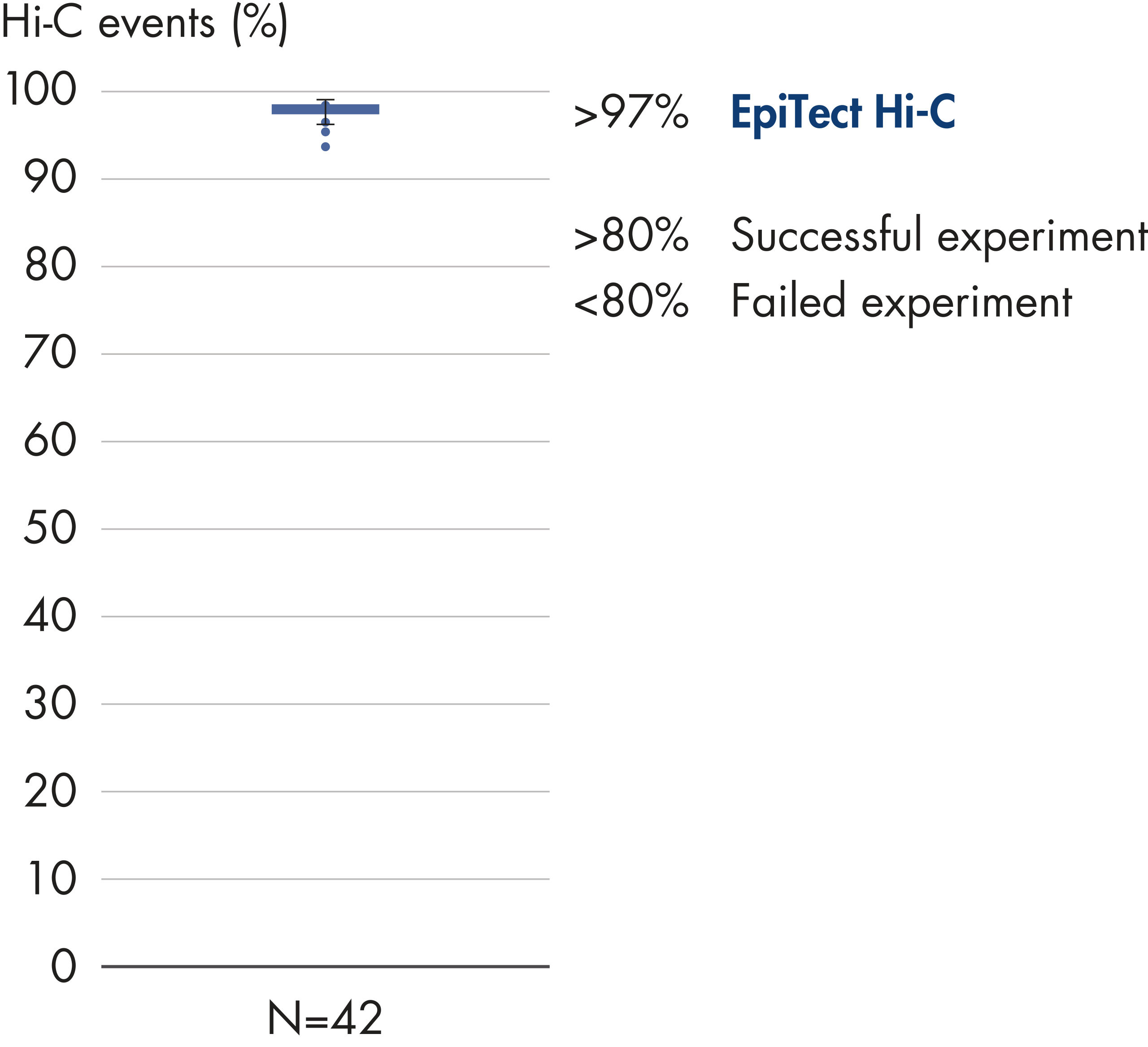
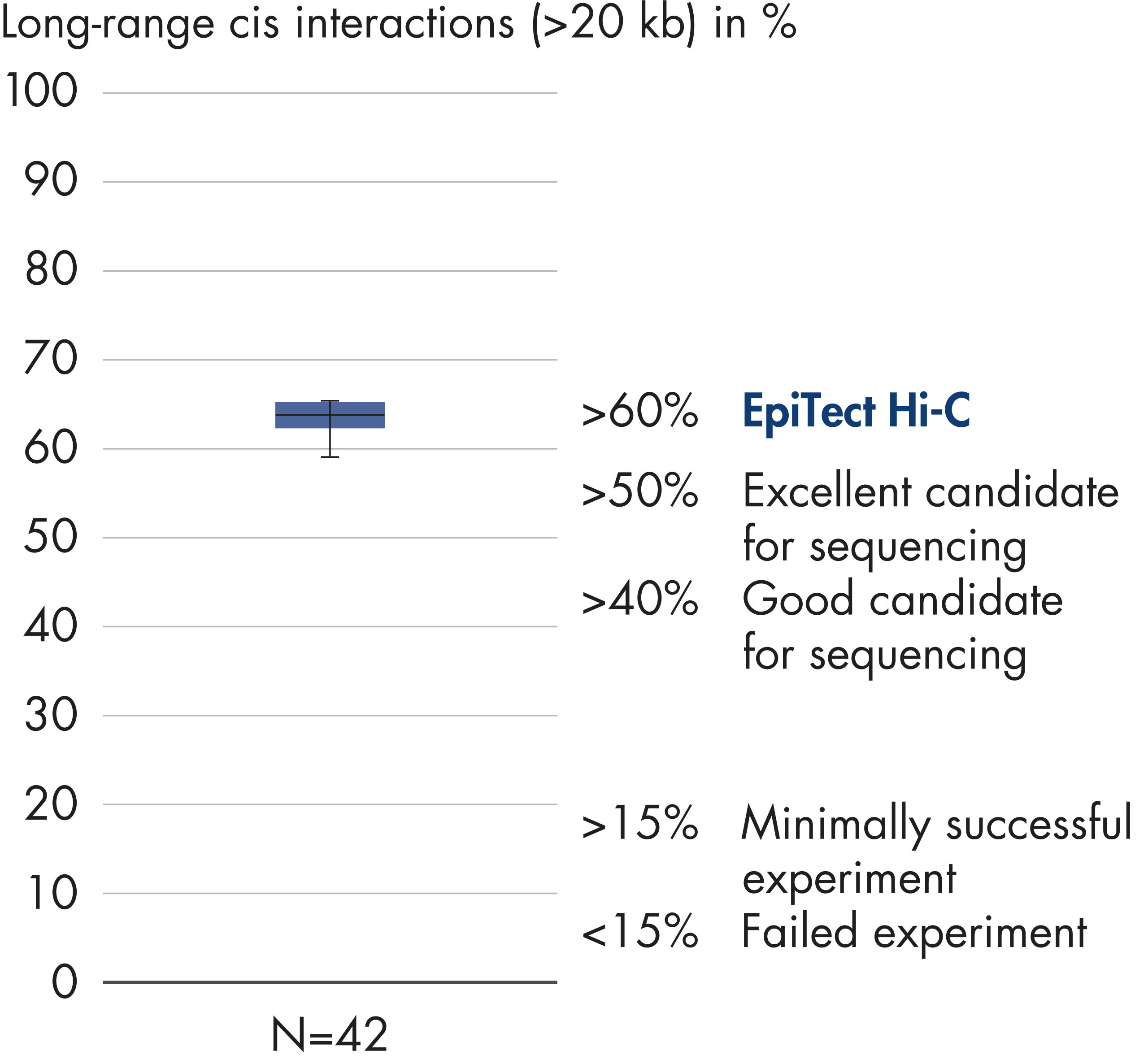
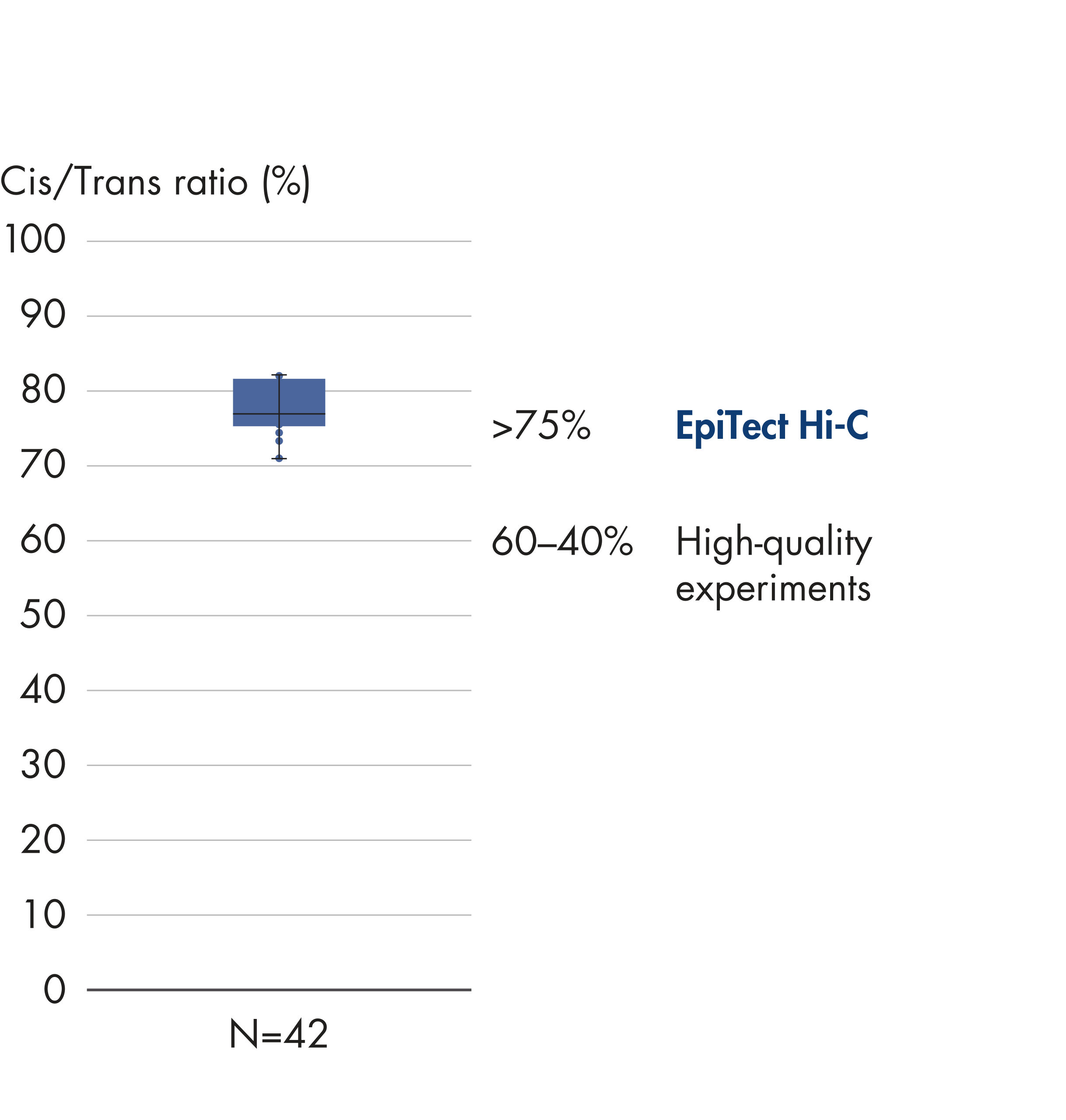
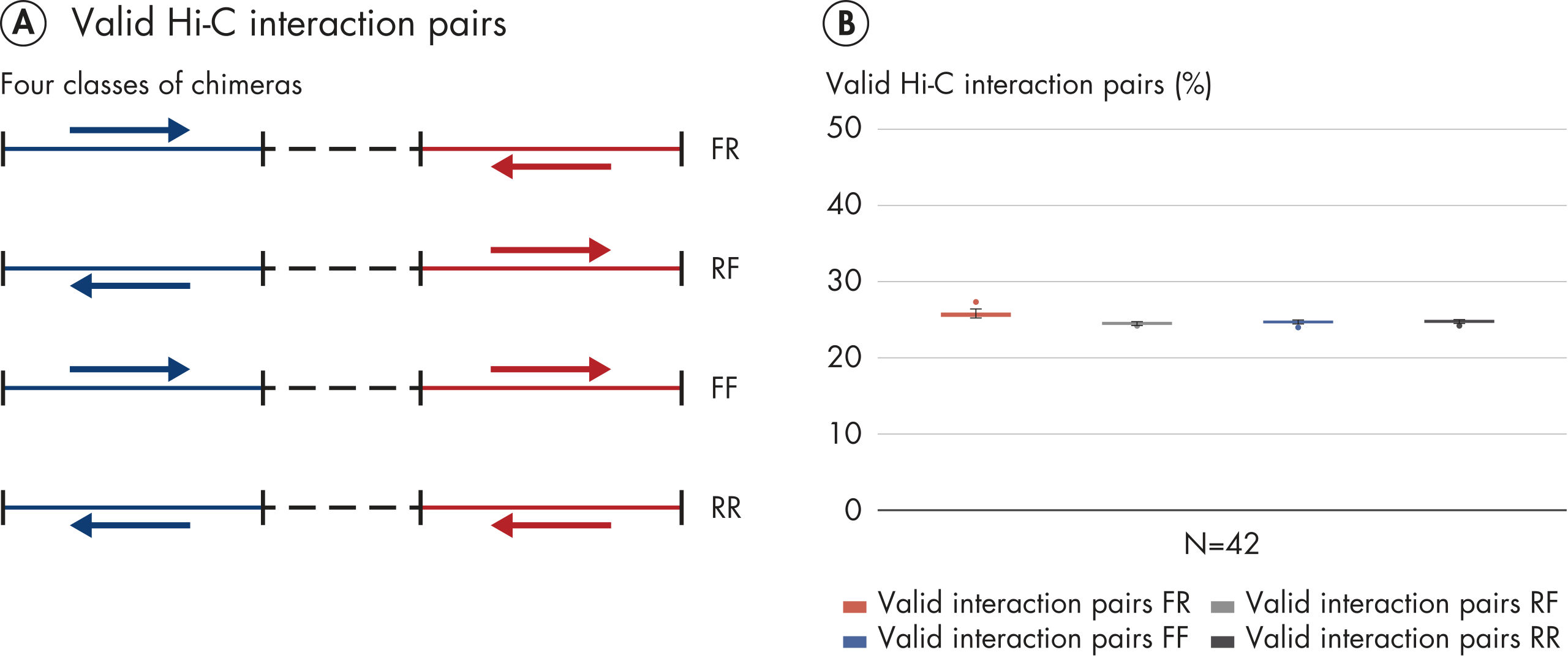
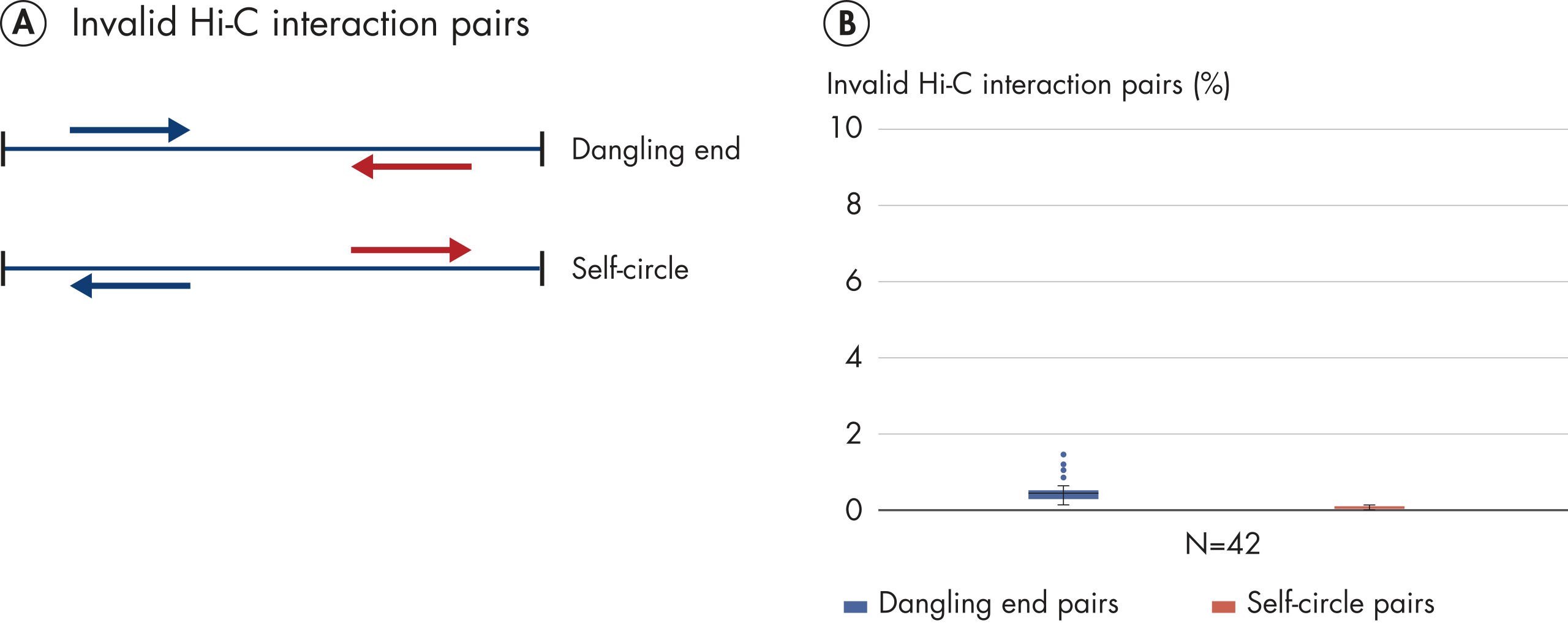
EpiTect Hi-C Kit 能生成高质量的 Hi-C NGS 文库,从而确保从高成本的下游深度测序中生成一流的数据。对来自 40 多个 EpiTect Hi-C 文库的测序结果进行了分析,以评估该试剂盒的性能。最重要的 QC 指标如下图所示: Hi-C 百分比、 长程顺式相互作用的百分比、 顺式/反式比率、 使用 EpiTect Hi-C Kit 不会产生链方向性偏差和 源自单一限制性片段的双端reads百分比。数据显示,平均而言,由 EpiTect Hi-C Kit 生成的 NGS 文库远超通常被视为足以成功完成 Hi-C 实验的标准。
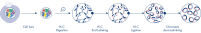
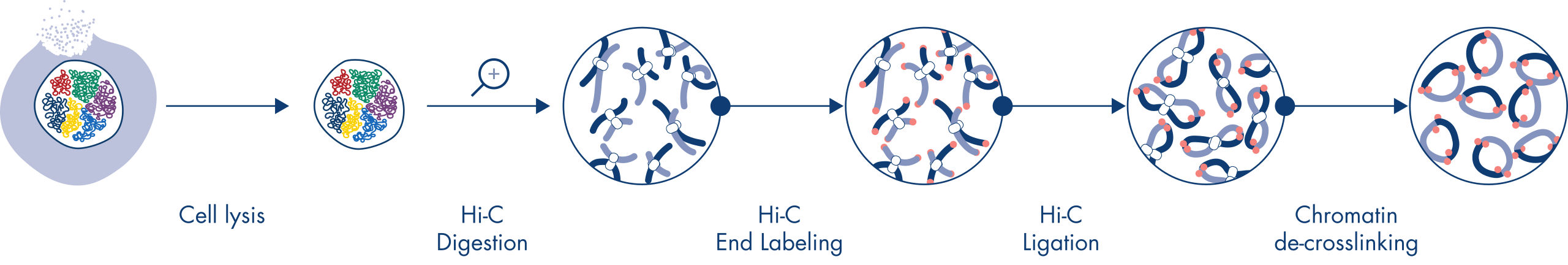
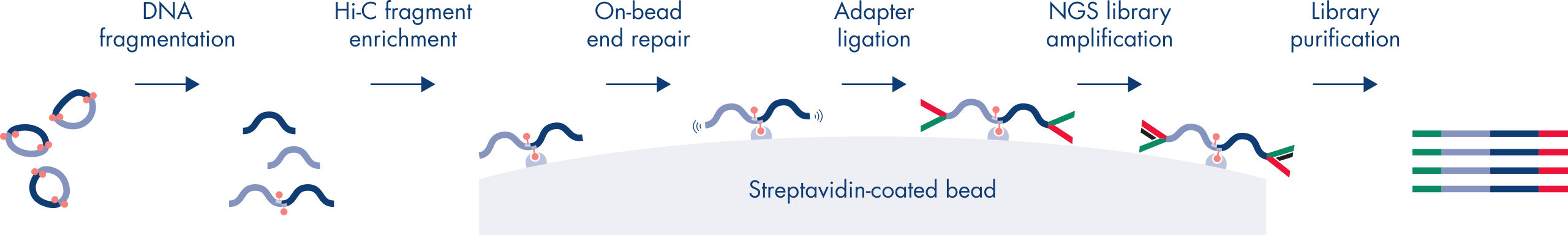
Hi-C 是一种邻位连接检测,可捕捉全基因组范围内的染色质相互作用。EpiTect Hi-C Kit 是一款专门用于生成与 Illumina 兼容的 NGS 文库(请参阅图 EpiTect Hi-C 工作流程 – 第 1 天和 EpiTect Hi-C 工作流程 – 第 2 天)的试剂盒。简言之,这项检测首先要纯化细胞核,其中的染色质构象已通过 DNA 结合蛋白和 DNA 的化学交联固定。然后用 4 bp 限制性内切酶完全消化 DNA。用生物素标记开放的 DNA 末端,然后进行连接。对 Hi-C 连接产物进行双端测序可识别出大量嵌合序列,这些序列来自空间中紧密相关的 DNA 链。两个序列连接在一起的概率与它们在空间中的平均距离有关。对连接接头进行定量,可以确定 DNA 的连接频率,并据此绘制染色质折叠的高分辨率图谱。
与已发表的方案相比,EpiTect Hi-C 工作流程(请参阅图 EpiTect Hi-C 工作流程 – 第 1 天和 EpiTect Hi-C 工作流程 – 第 2 天)有了明显的改进。长达一周的复杂程序已转变为只需 1.5 天的简单稳健程序。此外,样本投入要求降低了一个数量级,只需 5000 个细胞即可创建 Hi-C NGS 文库。该方案专为使用来自哺乳动物细胞培养物的交联细胞的工作而开发。
EpiTect Hi-C 实验流程是原位(即细胞核内)Hi-C 方法的一个版本,在该版本中,细胞核被温和地纯化和渗透,以便在最初的消化和连接步骤中保持基因组的空间组织。这一过程至关重要,可尽量减少因不能反映基因组结构的无义连接事件而产生的背景噪声。这是因为完好的细胞核限制了交联复合物的移动和随机碰撞,使得连接事件主要发生在拓扑关联的 DNA 片段之间。
构建 Hi-C NGS 文库
EpiTect Hi-C Kit 工作流程由 2 部分组成,每部分可在一天内完成。以下表格总结了方案的步骤,图 EpiTect Hi-C 工作流程 – 第 1 天和 EpiTect Hi-C 工作流程 – 第 2 天对这些步骤做了直观呈现。随附 Illumina 接头具有标签序列,可对多达 6 个样本进行并行测序。
要查看完整的方案,请参阅我们详细的 EpiTect Hi-C 手册。
数据分析
我们的 GeneGlobe 数据分析中心提供 Hi-C 数据分析。可使用 EpiTect Hi-C 数据分析门户分析 Hi-C 测序结果,该门户使用开源工具提供 QC 测序报告、Hi-C 连接矩阵和染色质连接图可视化。有关更多信息,请参阅我们的 EpiTect Hi-C 数据分析门户用户指南。
染色质构象
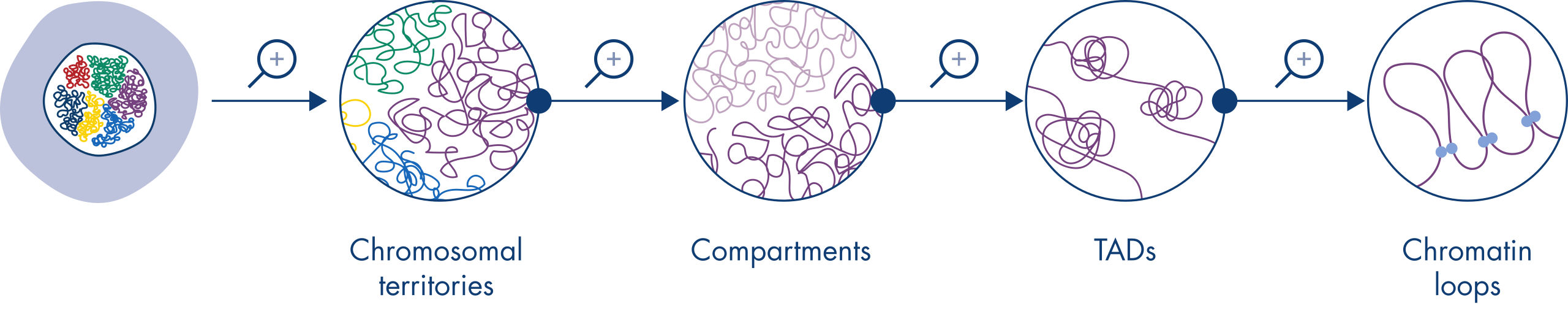
Hi-C 已迅速成为分析核组织的重要工具。对 Hi-C 数据的分析揭示了基因组结构惊人的复杂性,其中有多层空间组织将基因组划分为染色体疆域、染色体亚区室、拓扑相关结构域 (Topologically Associated Domain, TAD) 和分辨率不断提高的 DNA 环(请参阅图 染色质结构层级)。此外,基因组的组织是动态的,在发育过程中会发生变化。没有任何两种细胞类型的染色体折叠方式是相同的。
染色体重排和拷贝数变异
单个染色体在物理上被分隔成不同的领域,因此,Hi-C 捕捉到的 DNA 相互作用主要发生在同一染色体的 DNA 之间(顺式),染色体之间(反式)的相互作用很少。由于这种现象,Hi-C 可用作全基因组检测,以鉴定易位和其他值得关注的结构变异。与其他 NGS 技术相比,Hi-C 的覆盖范围要求极低,可以节省成本。此外,与标准的 NGS 方法相比,使用 Hi-C 可以更有效地检测涉及可映射性差区域的重排。方便的是,同样的 Hi-C 数据也可用于检测拷贝数变化。
基因组组装 – 单倍体分型
在对新物种的基因组进行测序和组装时,生成序列 scaffolds 往往会受到超出测序范围的大段重复序列的限制。在 Hi-C 数据中,绝大多数相互作用顺式发生在同一条染色体上的基因座之间。此外,这些顺式相互作用很大一部分间隔距离较长,发生在由数百万个 DNA 碱基间隔开的基因座之间。染色质相互作用的这些特性可用于通过排序、定向和连接使序列 scaffolds 成为近全长染色体,而无需参考基因组。利用同样的原理,Hi-C 交互作用图可用于通过将遗传变异分配给父系和母系姐妹染色体来创建二倍体基因组(请参阅图 Hi-C 测序数据的下游应用)。