RT2 Profiler PCR Arrays
信頼性と感度の高い遺伝子発現プロファイリング
信頼性と感度の高い遺伝子発現プロファイリング
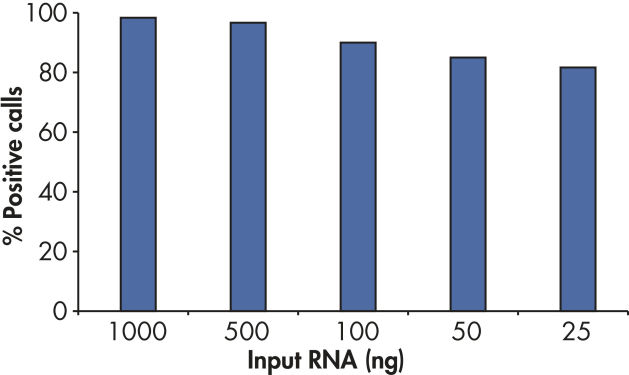
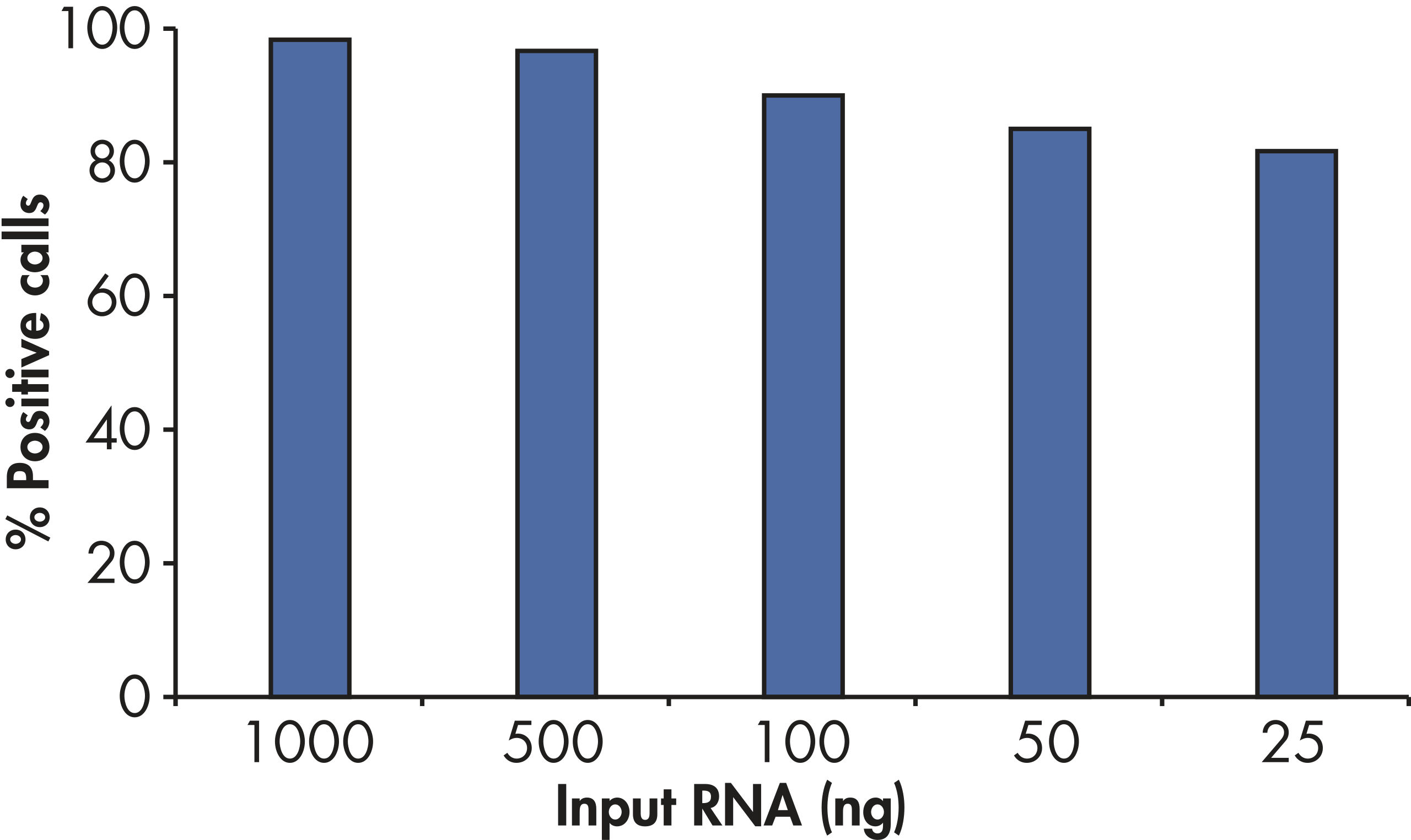
RT2 First Strand Kitの感度により、アレイプレートあたりわずか1 ngまたは最高5 µgのトータルRNAで80%を超えるpresent call rateが得られます(図“ わずか25 ngのRNAでポジティブな結果”)。
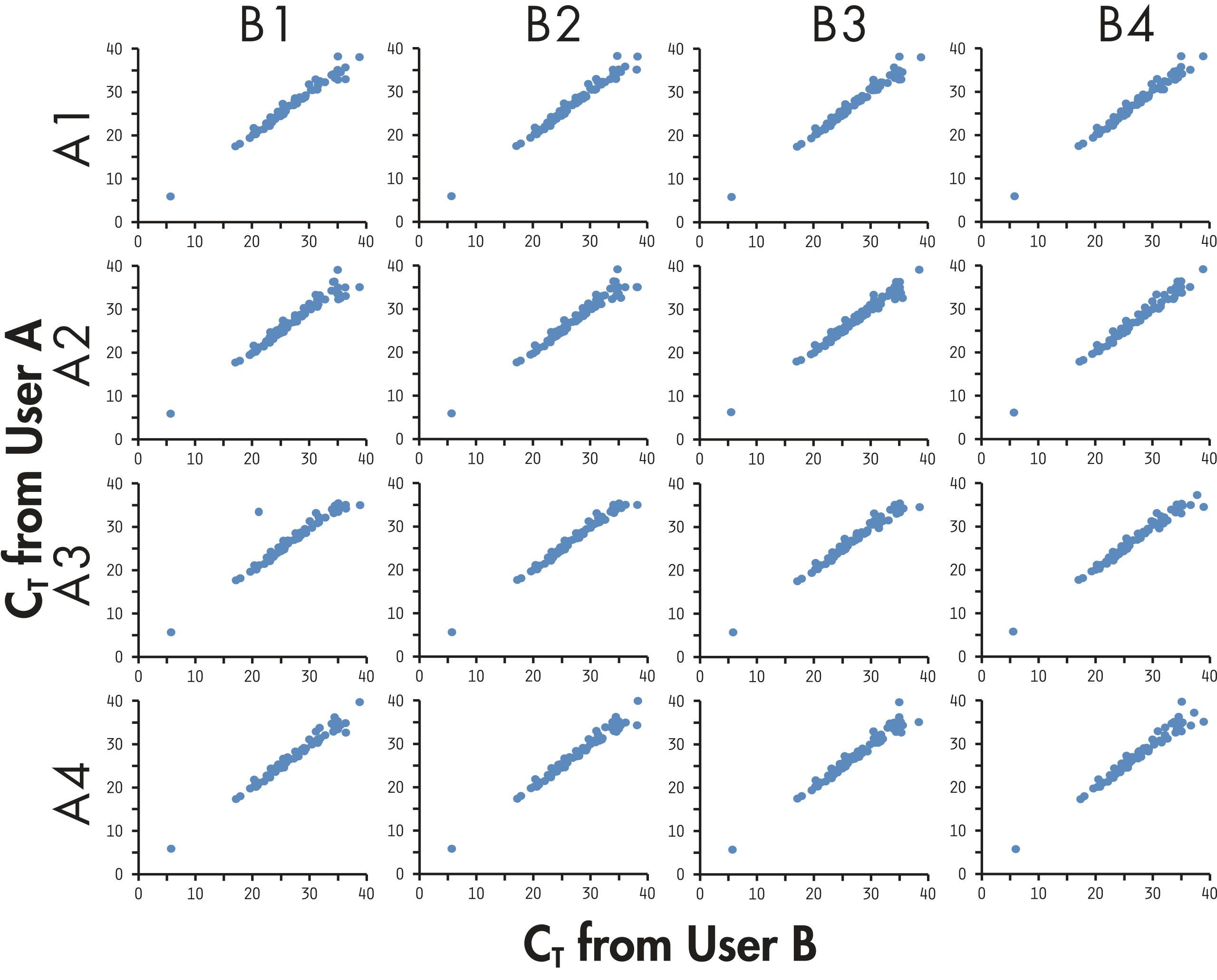
PCRアレイトータルシステムは、技術的なreplicate実験、異なるロット間、異なる装置間で高い相関性(相関係数が0.99を超える)をもつことが証明され、生体サンプルの発現の差を高い信頼性で検出できます(図“ 異なるユーザー間で高い再現性”)。
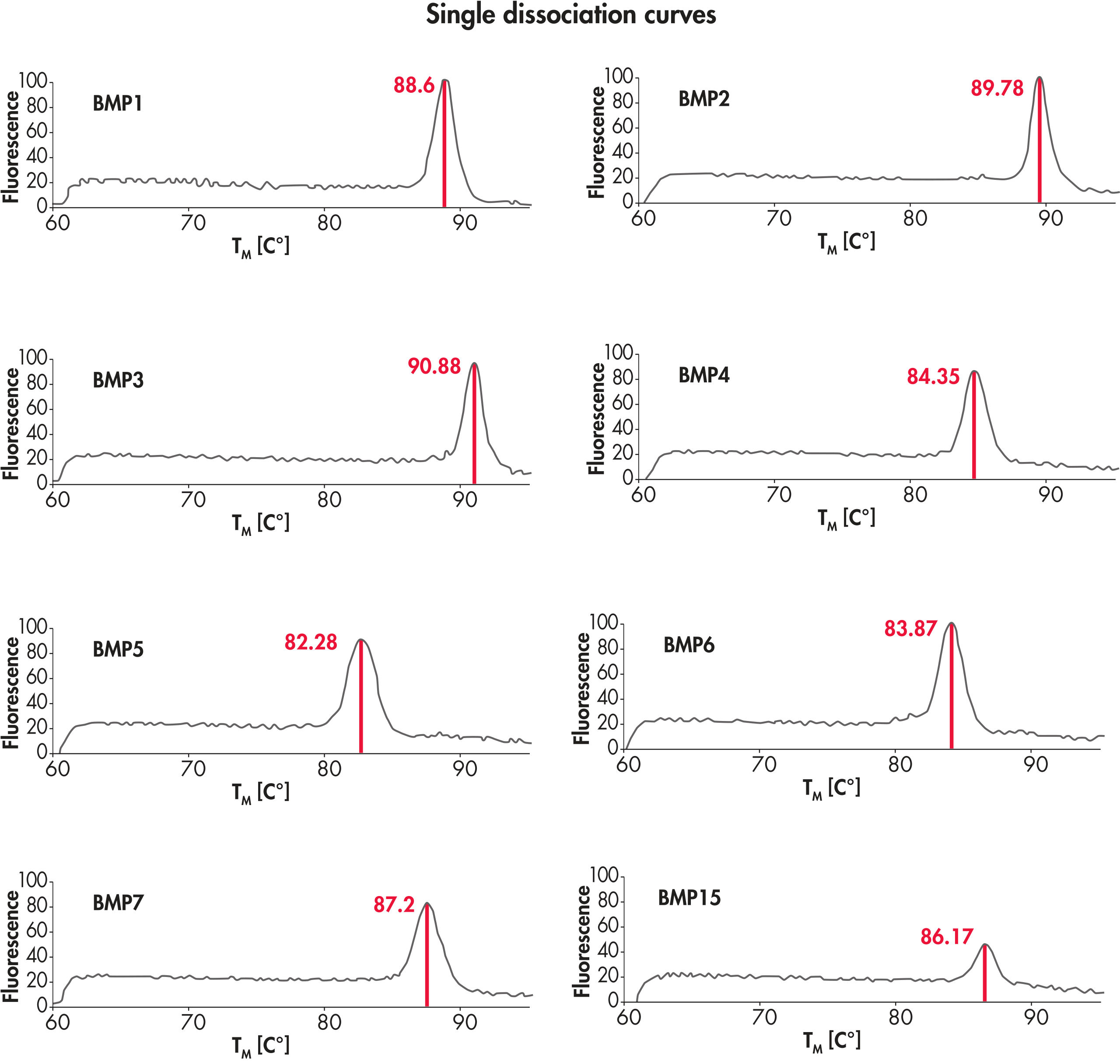
高品質のスタートRNAを用いたPCRアレイシステムでは、プライマーダイマーや二次産物がなく、予想されたサイズの単一バンドが得られるため、高精度なリアルタイムPCR 結果を実現します(図“ すべての反応で単一の遺伝子特異的産物”)。
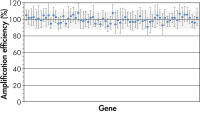
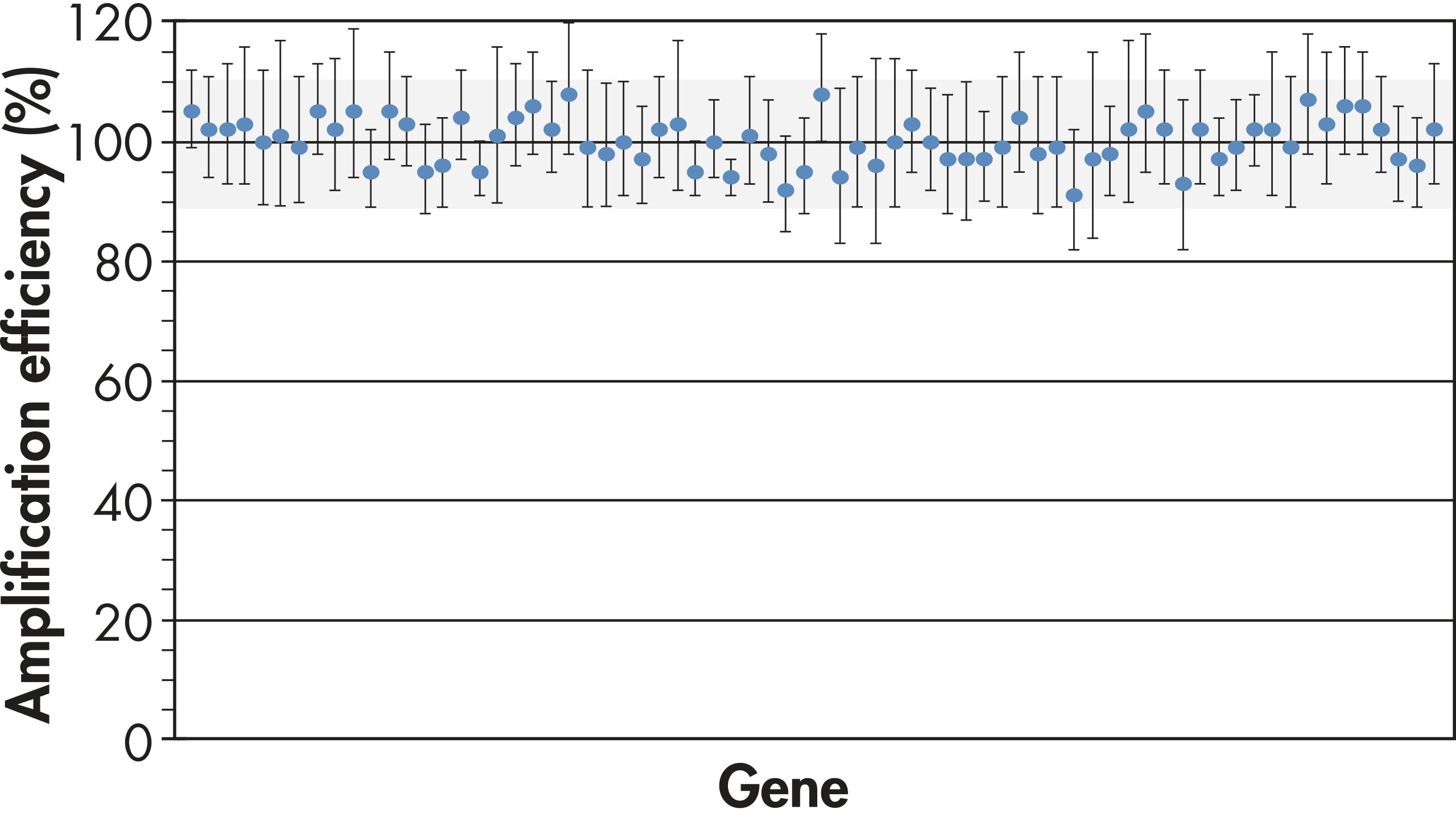
全遺伝子および全サンプルを通じて遺伝子発現レベルの正確な比較を行なうためには、一定したPCR増幅効率がPCRアレイテクノロジーに必須です。当社独自のプライマー設計アルゴリズムと、全プライマーアッセイの厳密なテストにより、PCRアレイでの全プライマーアッセイの高いパフォーマンスを保証します(図“ PCRアレイは高精度な結果を実現”)。
RT2 Profiler PCR Arrayは、フォーカスした遺伝子パネルの発現解析のための信頼できるツールです。各96ウエルプレート、384ウエルプレート、100-well disc PCR Arrayは、徹底的に研究されたパスウエイや疾病に関連した遺伝子パネル用のプライマーアッセイ(SYBR® Greenに至適化済み)を提供します。RT2 Profiler PCR Arrayはまたお客様の研究目的に即した遺伝子パネルをご提供するためにカスタマイズも可能です。高品質なプライマー設計とRT2 SYBR® Green qPCR Mastermix成分により、PCR アレイは同一サイクリング条件下で、96あるいは384種類の遺伝子特異的産物の同時増幅を実現します。
この組み合わせにより、RT2 Profiler PCR Arrayで正確なリアルタイムSYBR® Green結果に必要な高い感度と増幅効率が得られます。PCRアレイはどのような研究室でも容易に使用できます。
RT2 Profiler PCR Arrayは通常のサンプル(0.1~5 µgのRNA)、 FFPEサンプル、少量のサンプル(1~100 ngのRNA)から調製したRNAを使用して十分な感度を発揮できます。
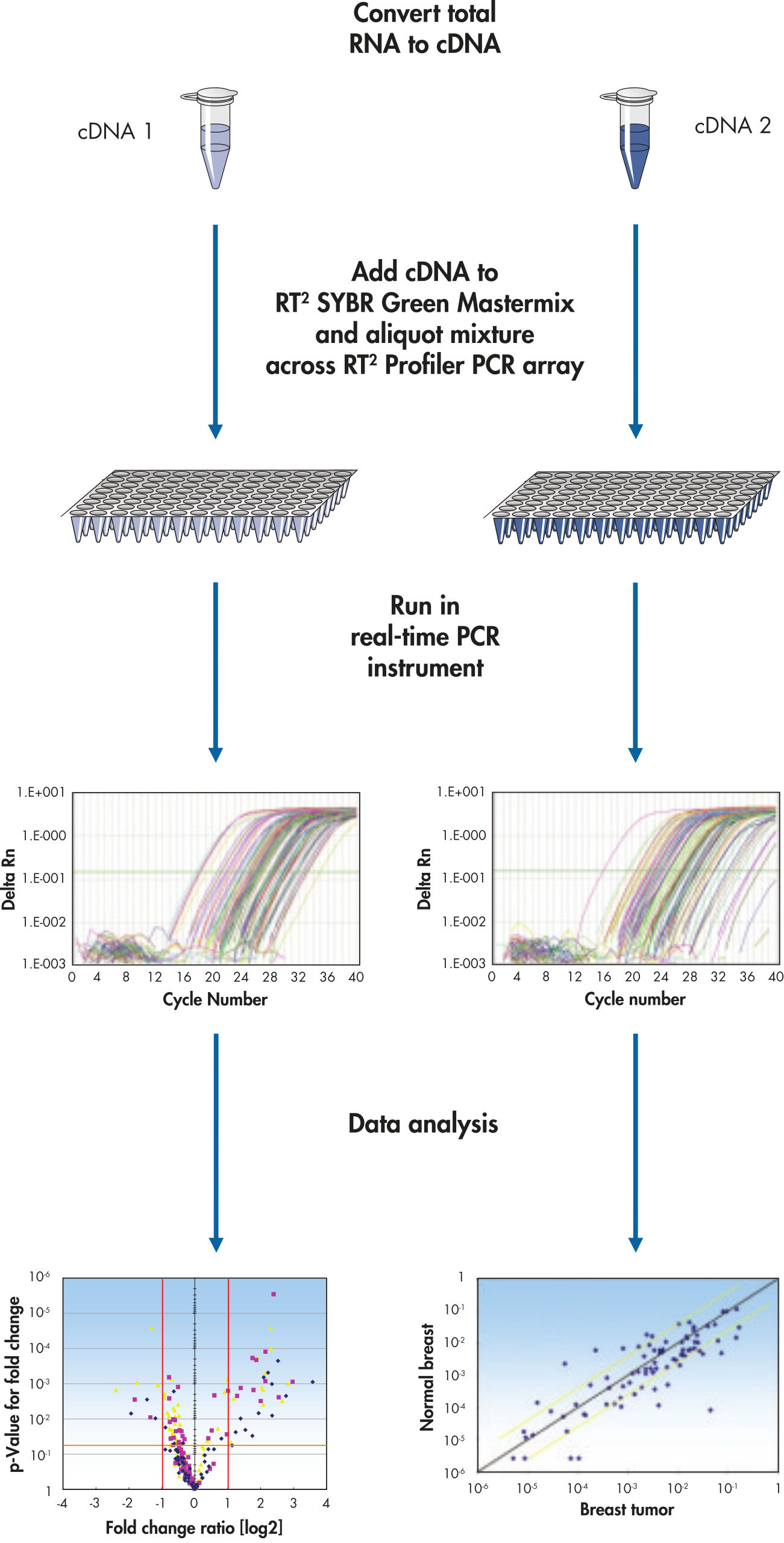
cDNAテンプレートと適切で即使用可能なPCRマスターミックスを混和し、同一プレートの各ウエルに等量分注し、リアルタイムPCRサイクリングプリグラムを起動します(フローチャート“ 簡単な操作”)。RT2 Profiler PCR Arrayは、QIAGEN、ABI、Bio-Rad、Eppendorf、Roche、Stratageneの装置で使用可能です。
RT2 Profiler PCR Arrayは96ウエルプレート、384ウエルプレート、100-well discフォーマットで入手可能で、84あるいは370種類の疾病あるいはパスウエイに関連する遺伝子と5種類のハウスキーピング遺伝子の発現を調べることができます。RT2 Profiler PCR Arrayには以下の目的のためにコントロールが入っています:
エクセルベースのデータ解析テンプレートあるいはウエブベースのソフトウェアを用いてデータ解析を簡単に行なえます。データ解析はΔΔCT法に基づき、ハウスキーピング遺伝子に対して生データを標準化します。
RT2 PCR Profiler Arrayは次のような生物および医学研究領域で使用できます。