EpiTect Hi-C Kit
Para el mapeo de alta resolucion para plegamiento de cromatina, ensamblaje de alta calidad de secuencias genomicas, ajuste de haplotipos e identif. de reordenamientos cromosomicos.
Para el mapeo de alta resolucion para plegamiento de cromatina, ensamblaje de alta calidad de secuencias genomicas, ajuste de haplotipos e identif. de reordenamientos cromosomicos.
✓ Procesamiento automático sin interrupción de pedidos en línea
✓ Servicio técnico y para productos experto y profesional
✓ Realización y repetición de pedidos rápidas y fiables
Cat. No. / ID: 59971
✓ Procesamiento automático sin interrupción de pedidos en línea
✓ Servicio técnico y para productos experto y profesional
✓ Realización y repetición de pedidos rápidas y fiables
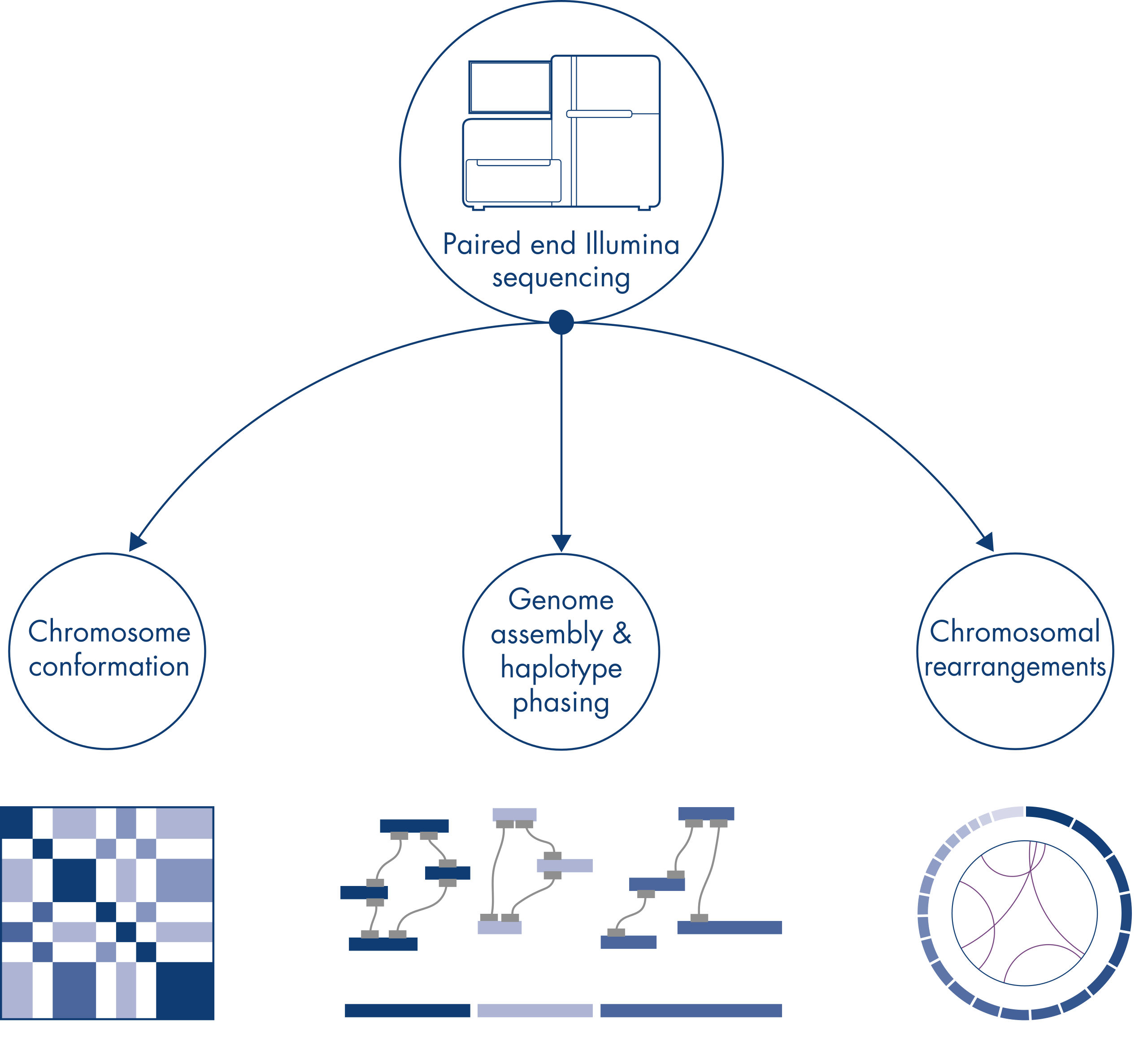
El Hi-C se diseñó originalmente como una potente técnica para la captura de la estructura cromosómica en todo el genoma, lo que permitía la caracterización del plegamiento de la cromatina con una resolución en kb. Sin embargo, esta técnica también tiene otras aplicaciones relevantes. Por ejemplo, el Hi-C se utiliza para generar ensamblajes genómicos altamente contiguos, con pocos armazones y muy largos, a partir de organismos sin un genoma de referencia conocido. Además, el Hi-C es muy útil para el ajuste de haplotipos y la identificación de reordenamientos cromosómicos.
El EpiTect Hi-C Kit ofrece un protocolo robusto pero sencillo y rápido, con pocos requisitos de entrada de células que permite la generación de bibliotecas de NGS Illumina Hi-C de alta calidad a partir de células entrecruzadas en menos de 2 días.
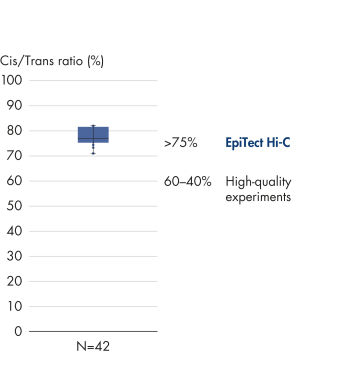
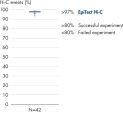
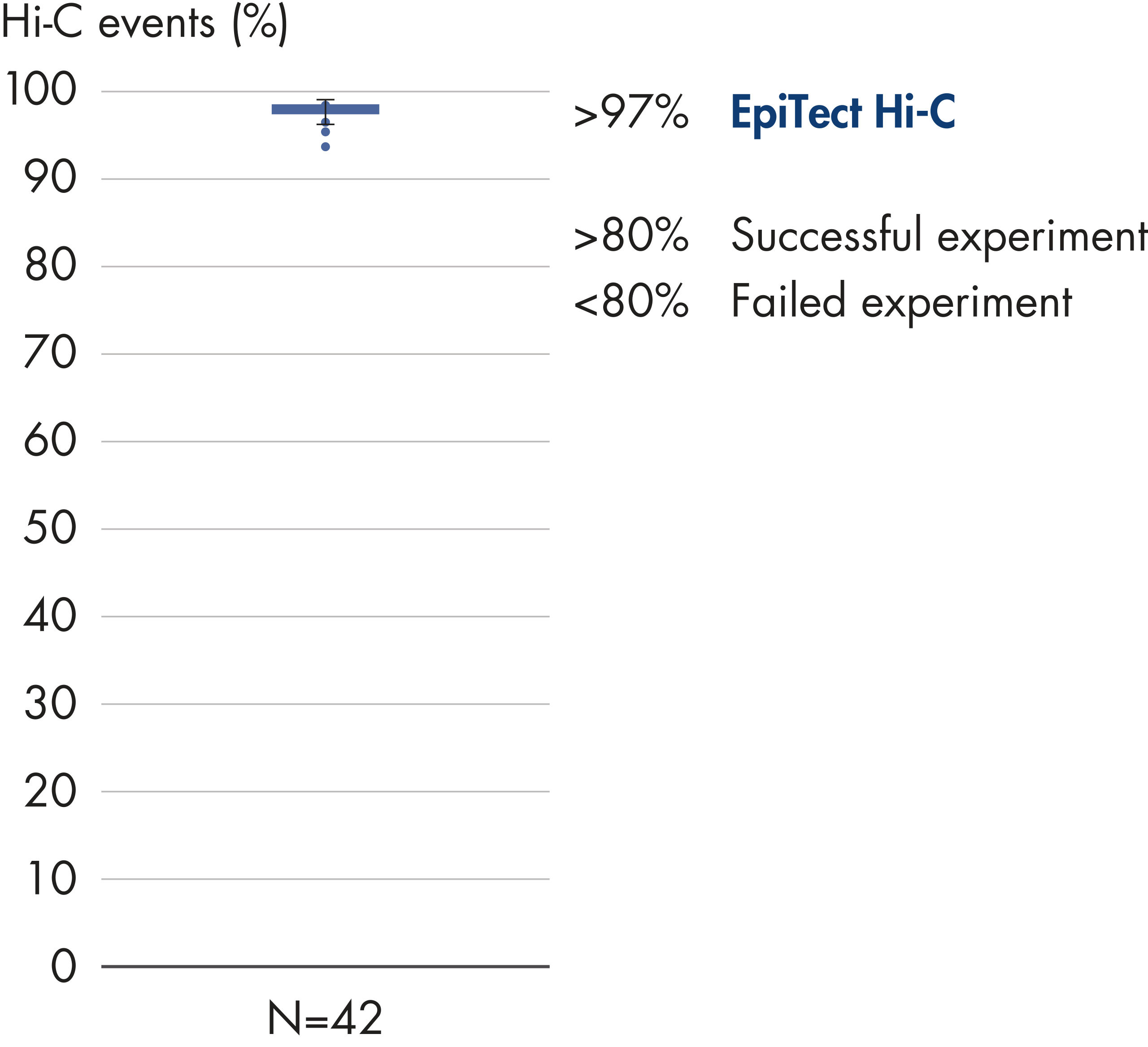
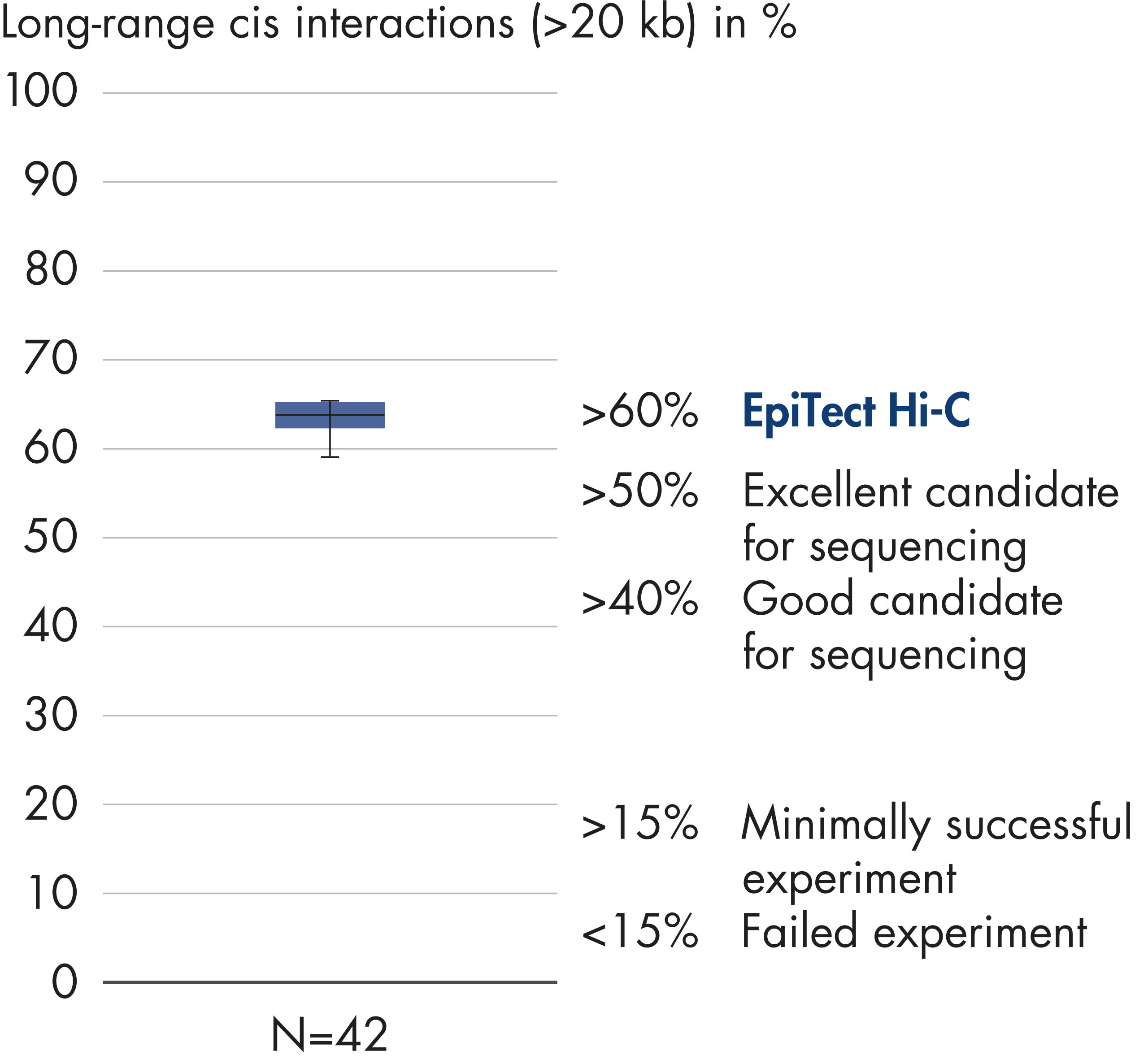
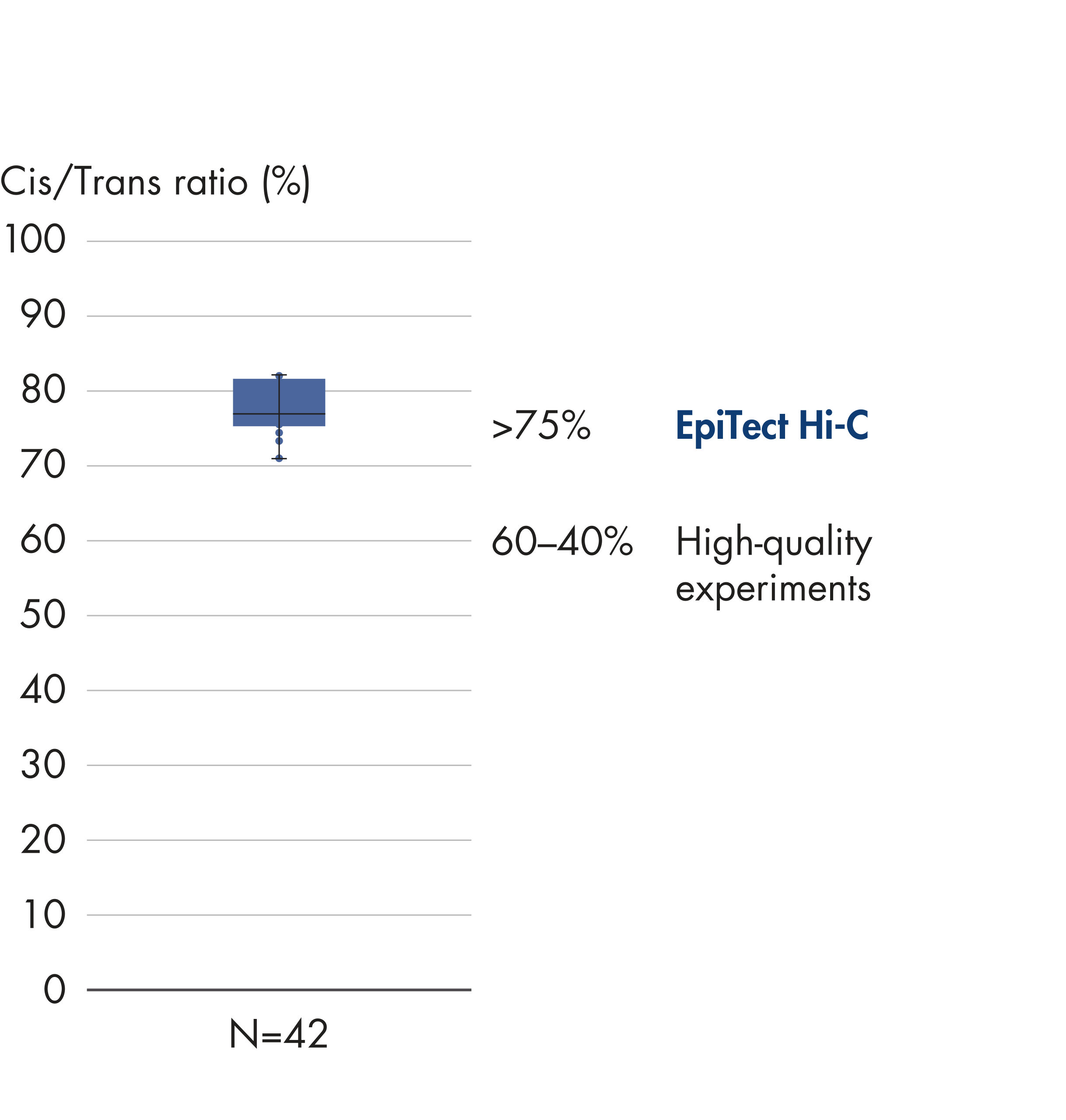
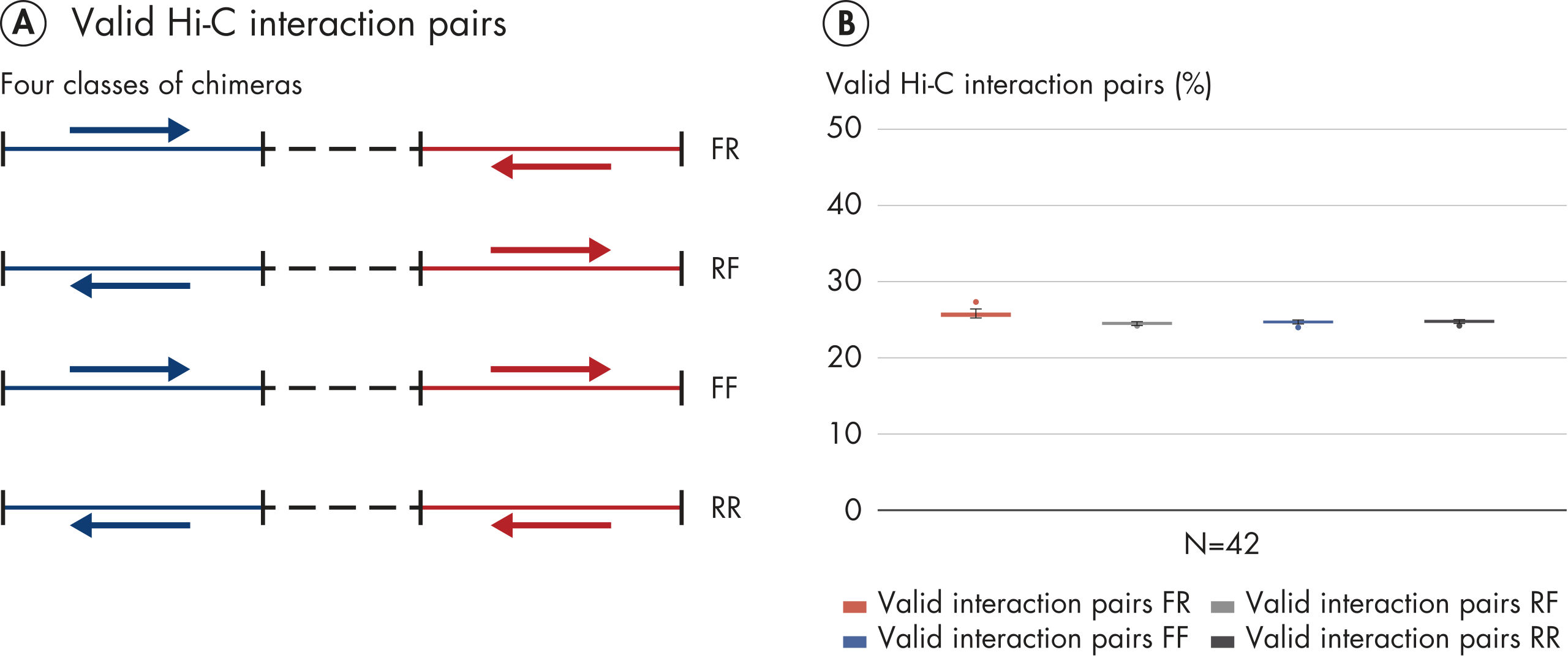
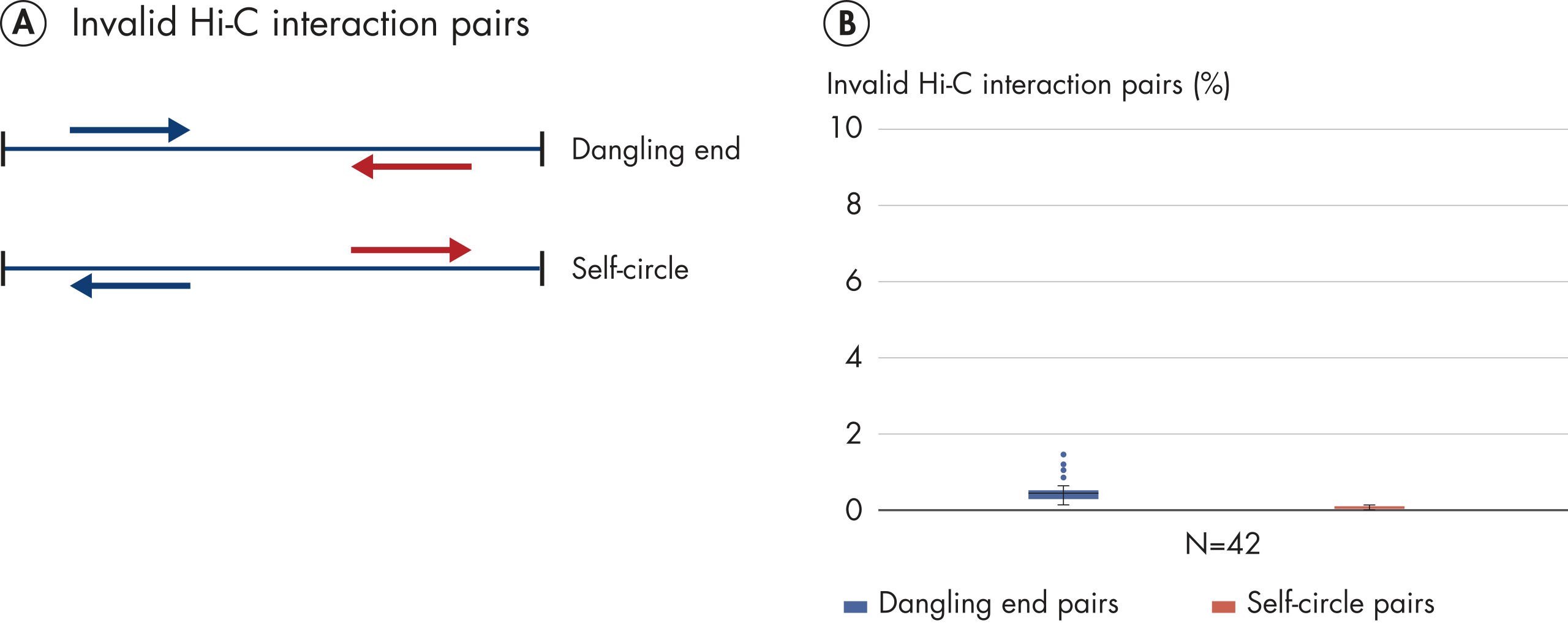
El EpiTect Hi-C Kit proporciona bibliotecas Hi-C de NGS de alta calidad, lo que garantiza la generación de datos de primera calidad a partir de la costosa secuenciación posterior exhaustiva. Se han analizado los resultados de secuenciación de más de 40 bibliotecas EpiTect Hi-C para evaluar el rendimiento del kit. Las medidas de control de calidad más importantes se muestran en las figuras siguientes: Porcentaje de sucesos Hi-C, Porcentaje de interacciones cis de largo alcance, Relación Cis/Trans, Sin sesgo de orientación de la hebra con el EpiTect Hi-C Kit y Porcentaje de lecturas emparejadas derivadas de un único fragmento de restricción. Los datos muestran que el EpiTect Hi-C Kit genera bibliotecas de NGS que, de media, superan con creces los criterios que normalmente se consideran suficientes para un experimento de Hi-C satisfactorio.
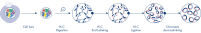
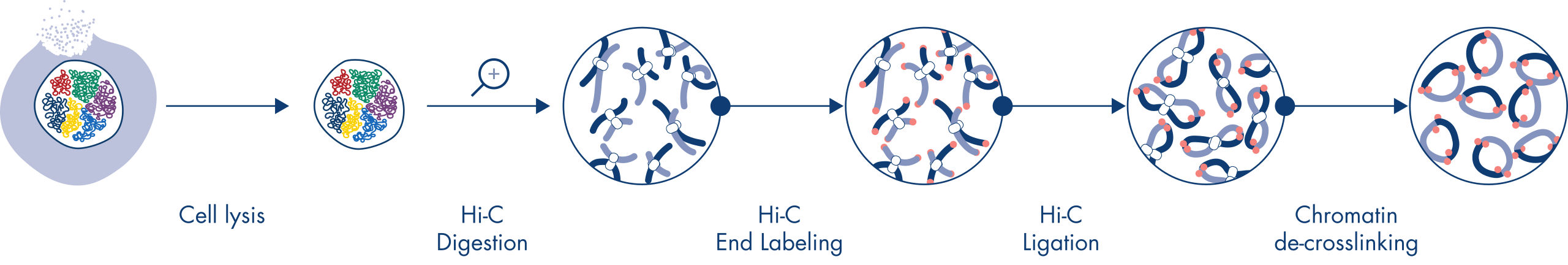
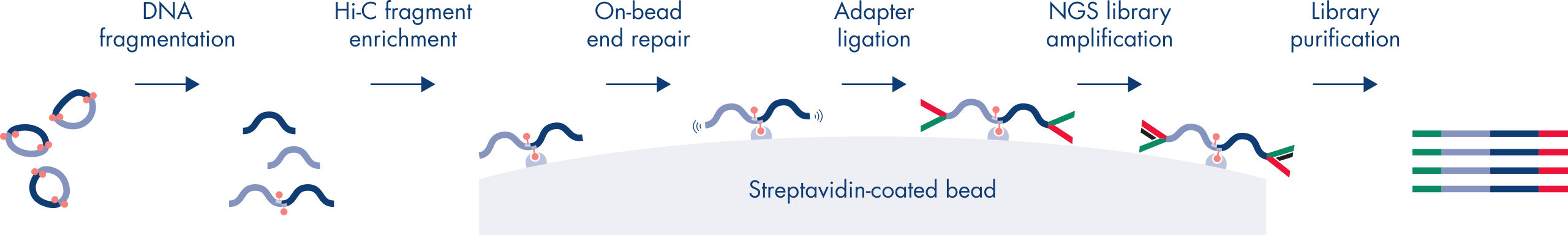
Hi-C es un ensayo de unión de proximidad que captura las interacciones de la cromatina a escala de genoma completo. El EpiTect Hi-C Kit es una preparación de ADN especializada que da como resultado una biblioteca de NGS compatible con Illumina (consulte las figuras Flujo de trabajo de EpiTect Hi-C Kit: día 1 y Flujo de trabajo de EpiTect Hi-C Kit: día 2). A grandes rasgos, el ensayo empieza con la purificación de los núcleos, en los que la estructura de la cromatina se ha congelado mediante reticulación química de las proteínas de unión de ADN y el ADN. A continuación, el ADN se hidroliza completamente con una enzima de restricción de 4 bp. Los extremos abiertos del ADN se marcan con biotina y, posteriormente, se ligan. La secuenciación de ambos extremos de los productos de la ligación Hi-C identifica un gran número de secuencias mixtas derivadas de hebras de ADN que estaban estrechamente unidas en el espacio. La posibilidad de que dos secuencias acaben ligadas está en función de su distancia media en el espacio. La cuantificación de las uniones de ligadura permite determinar las frecuencias de contacto del ADN a partir de las cuales se puede lograr un mapeo de alta resolución del plegamiento de la cromatina.
El flujo de trabajo de EpiTect Hi-C Kit (consulte las figuras Flujo de trabajo de EpiTect Hi-C Kit: día 1 y Flujo de trabajo de EpiTect Hi-C Kit: día 2) representa una notable mejora con respecto a los protocolos publicados. Aquello que antes era un procedimiento complicado y que duraba una semana, ahora es un protocolo robusto y sencillo que solo necesita 1,5 días. Además, la necesidad de introducción de muestras se ha reducido en un orden de magnitud, lo que permite crear bibliotecas Hi-C de NGS a partir de tan solo 5000 células. El protocolo se ha desarrollado para trabajar con células reticuladas procedentes de cultivos celulares de mamíferos.
El procedimiento EpiTect Hi-C es una versión del método Hi-C in situ (es decir, en el núcleo) en el que los núcleos se permeabilizan y se purifican cuidadosamente para mantener la organización espacial del genoma durante los pasos iniciales a la hidrolización y ligadura. El proceso es fundamental para reducir el ruido de fondo de los sucesos de ligadura no informativos que no reflejan la organización genómica. Esto se debe a que los núcleos intactos limitan el movimiento y las colisiones aleatorias de los complejos reticulados, de modo que los sucesos de ligadura se producen predominantemente entre fragmentos de ADN topológicamente asociados.
Construcción de las bibliotecas Hi-C de NGS
El flujo de trabajo de EpiTect Hi-C Kit consiste en dos partes; cada una de ellas puede completarse en un día. Los pasos del protocolo se resumen en las tablas siguientes y se pueden ver en las figuras Flujo de trabajo de EpiTect Hi-C Kit: día 1 y Flujo de trabajo de EpiTect Hi-C Kit: día 2. Los adaptadores Illumina suministrados tienen secuencias de códigos de barras que permiten la secuenciación múltiple de hasta 6 muestras.
Para visualizar el protocolo completo, consulte el detallado Manual de uso del EpiTect Hi-C.
Análisis de los datos
El análisis de datos de Hi-C se proporciona en nuestro Centro de análisis de datos de GeneGlobe. Los resultados de las secuencias Hi-C se pueden analizar mediante el Portal de análisis de datos de EpiTect Hi-C, el cual utiliza herramientas de código abierto para proporcionar un informe de secuenciación de control de calidad, matrices de contactos Hi-C y visualización de mapas de contactos de cromatina. Para obtener más información, consulte nuestra Guía del usuario del portal de análisis de datos de EpiTect Hi-C.
Estructura de la cromatina
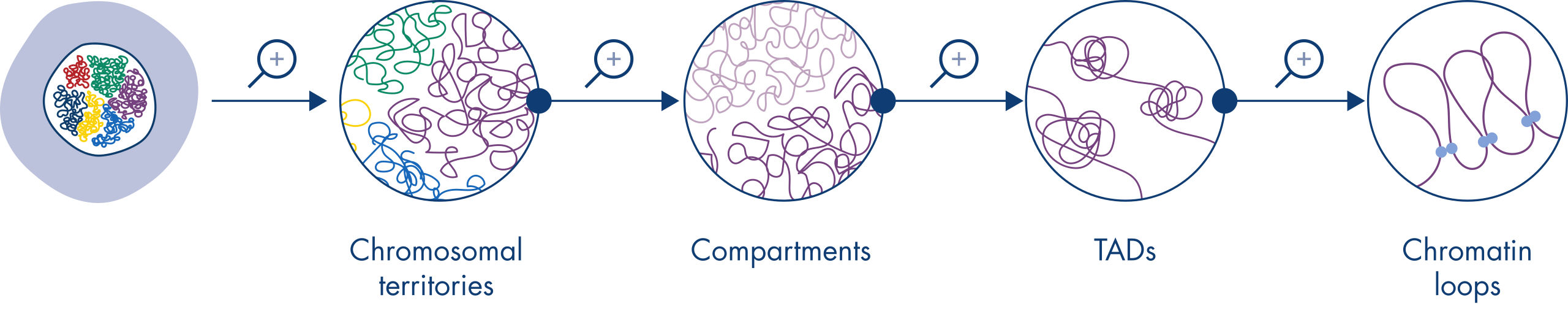
El Hi-C se ha convertido en una herramienta fundamental para el análisis de la organización nuclear. El análisis de los datos de Hi-C ha revelado la espectacular complejidad de la arquitectura genómica, con múltiples capas de organización espacial que dividen el genoma en territorios cromosómicos, subcompartimentos cromosómicos, dominios topológicamente asociados (TAD) y bucles del ADN a una resolución cada vez mayor (consulte la figura Niveles de organización de la cromatina). Además, la organización genómica es dinámica y cambia durante el desarrollo. No existen dos tipos de célula en que los cromosomas se plieguen de la misma manera.
Reordenamientos cromosómicos y variantes del número de copias
Los cromosomas individuales se separan físicamente en territorios diferenciados y, por lo tanto, las interacciones de ADN captadas por Hi-C tienen lugar principalmente entre el ADN del mismo cromosoma (en cis) con una escasa interacción entre cromosomas (en trans). Debido a este fenómeno, el Hi-C puede utilizarse como un ensayo de genoma completo para identificar translocaciones y otras variantes estructurales de interés. En comparación con otras técnicas de NGS, el Hi-C requiere una cobertura extremadamente baja, cosa que puede reducir los costes. Asimismo, con el Hi-C se pueden detectar mejor los reordenamientos que afectan a las regiones con mapeados deficientes que con los métodos de NGS estándares. Así, se pueden utilizar los mismos datos de Hi-C para detectar cambios en el número de copias.
Ensamblaje genómico: ajuste de haplotipos
Al secuenciar y ensamblar genomas de nuevas especies, la generación de armazones de secuencias suele verse limitada por grandes tramos de secuencias repetitivas que se extienden más allá del alcance de la secuenciación. En los datos de Hi-C, la gran mayoría de las interacciones se producen en cis entre locus del mismo cromosoma. Además, una parte considerable de estas interacciones cis es de largo alcance y se produce entre locus separados por millones de bases de ADN. Estas propiedades de las interacciones de la cromatina pueden aprovecharse para ordenar, orientar y unir armazones de secuencias en cromosomas casi completos sin necesidad de un cromosoma de referencia. De acuerdo con los mismos principios, los mapas de interacciones de Hi-C pueden utilizarse para crear genomas diploides mediante la asignación de variantes genéticas a los cromosomas homólogos materno y paterno (consulte la figura Aplicaciones posteriores de los datos de secuenciación de Hi-C).