EpiTect Hi-C Kit
Für die hochauflösende Kartierung der Chromatinfaltung, qualitativ hochwertige Assemblierung von Genomsequenzen, Identifizierung von chromosomalen Rearrangements und das Haplotyp-Phasing
Für die hochauflösende Kartierung der Chromatinfaltung, qualitativ hochwertige Assemblierung von Genomsequenzen, Identifizierung von chromosomalen Rearrangements und das Haplotyp-Phasing
✓ Automatische Verarbeitung von Online-Bestellungen 24/7
✓ Sachkundiger und professioneller technischer und Produkt-Support
✓ Schnelle und zuverlässige (Nach-)Bestellung
Cat. No. / ID: 59971
✓ Automatische Verarbeitung von Online-Bestellungen 24/7
✓ Sachkundiger und professioneller technischer und Produkt-Support
✓ Schnelle und zuverlässige (Nach-)Bestellung
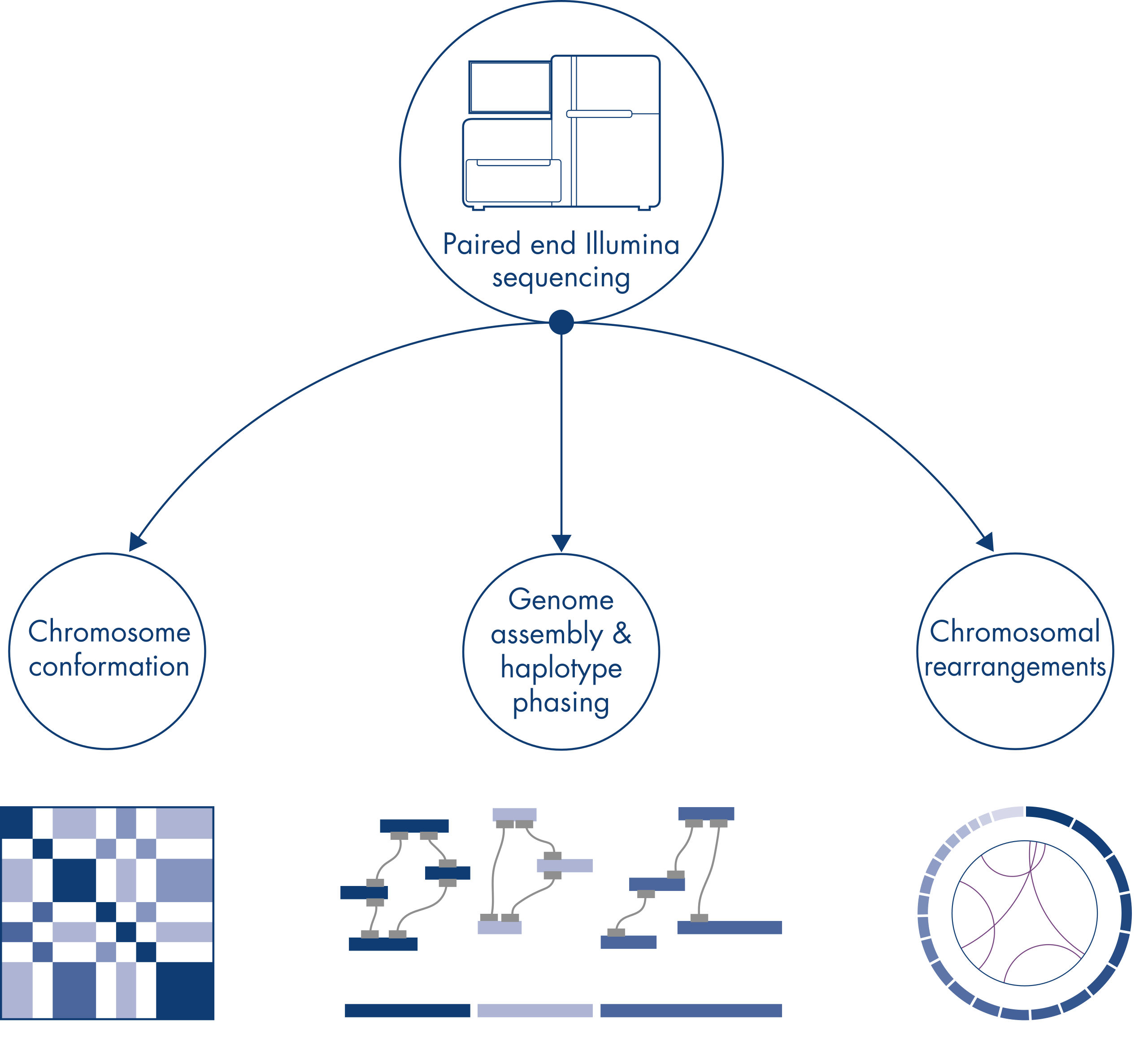
Hi-C wurde ursprünglich als leistungsstarke Technik zur genomweiten Erfassung der Chromosomenkonformation entwickelt, die die Charakterisierung der Chromatinfaltung mit kb-Auflösung ermöglicht. Die Technologie hat aber auch andere wichtige Anwendungen. So wird Hi-C beispielsweise zur Erstellung von hochgradig zusammenhängenden Genomassemblierungen mit wenigen und sehr langen Scaffolds aus Organismen ohne bekanntes Referenzgenom verwendet. Darüber hinaus ist Hi-C auch sehr nützlich für das Haplotyp-Phasing und den Nachweis von chromosomalen Rearrangements.
Das EpiTect Hi-C Kit beruht auf einem robusten, aber dennoch einfachen und schnellen Protokoll, das mit kleinen Ausgangsmengen an Zellmaterial durchgeführt werden kann. Es ermöglicht die Generierung von qualitativ hochwertigen Hi-C Illumina NGS-Bibliotheken aus quervernetzten Zellen in weniger als 2 Tagen.
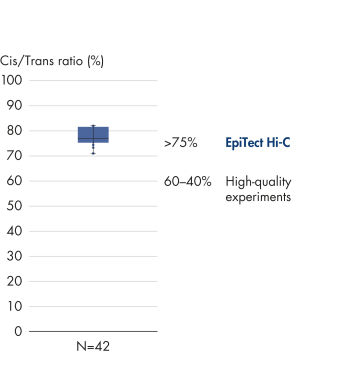
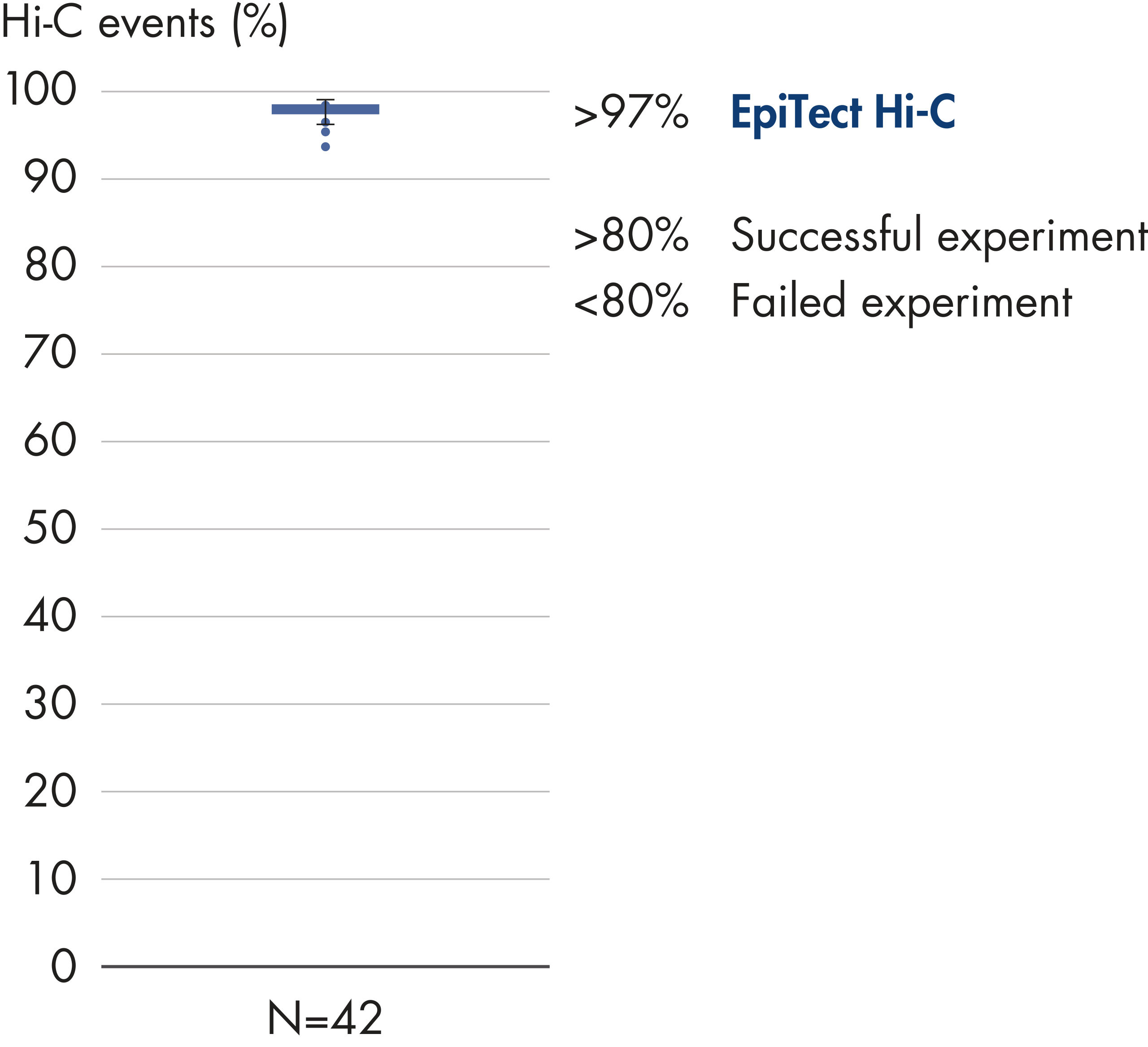
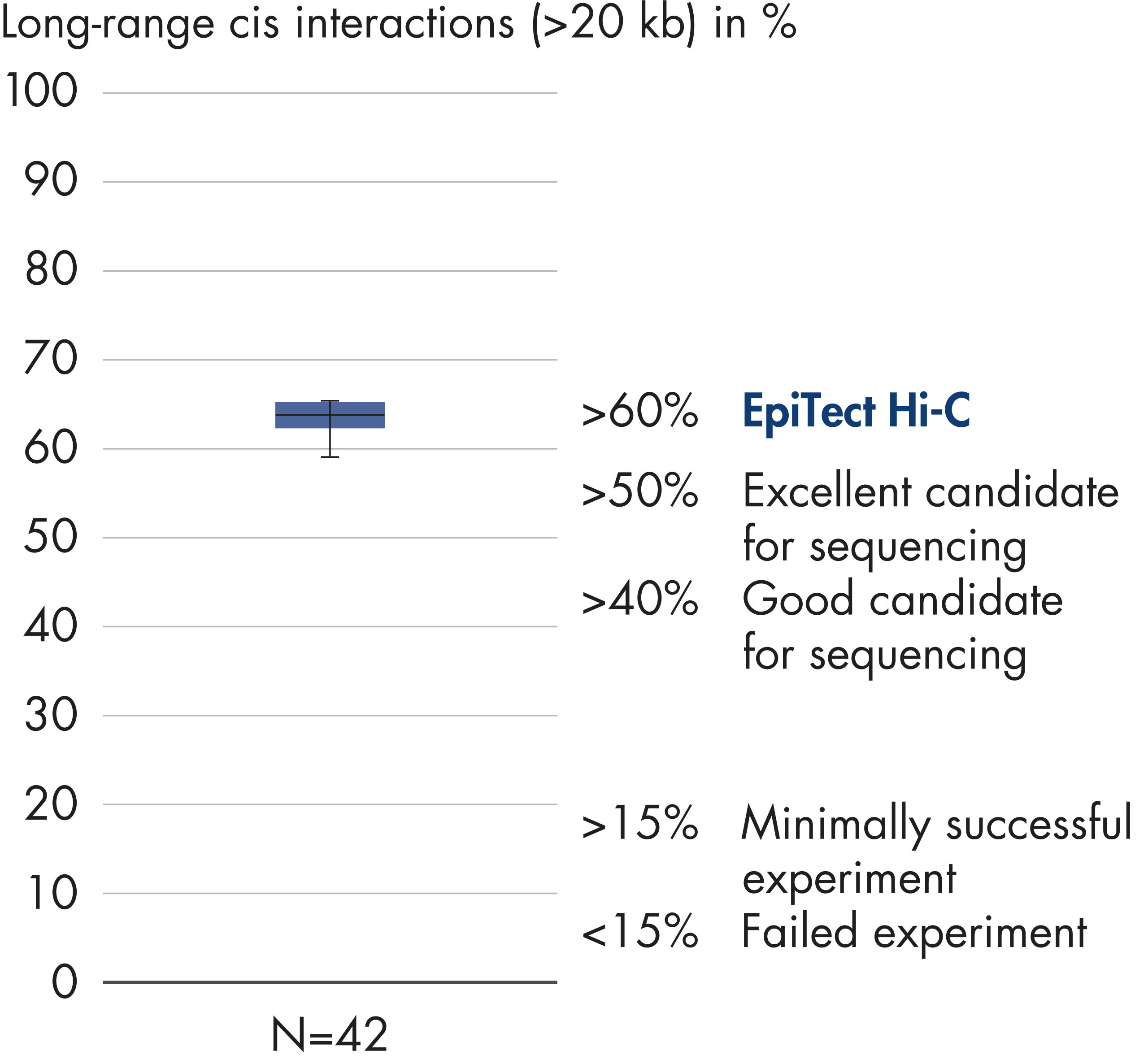
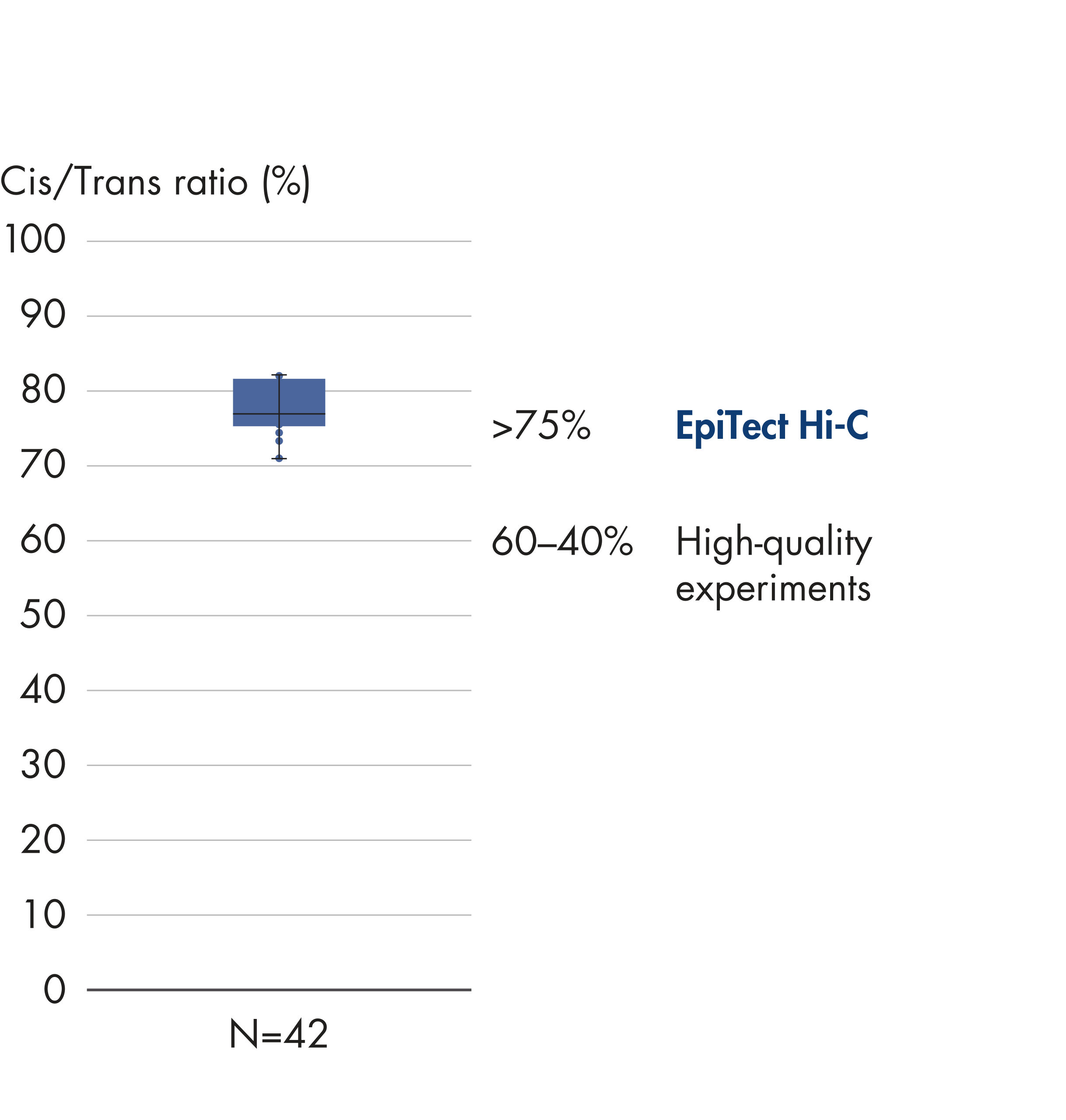
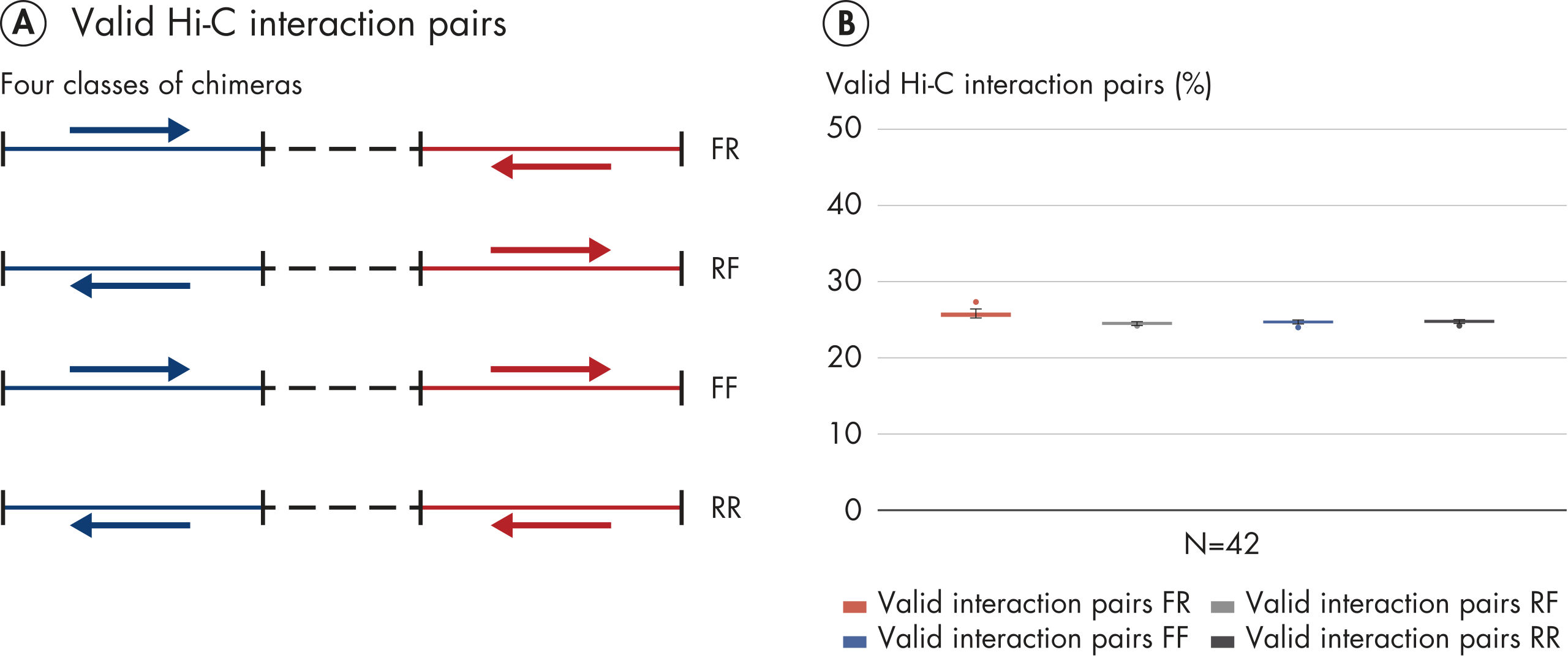
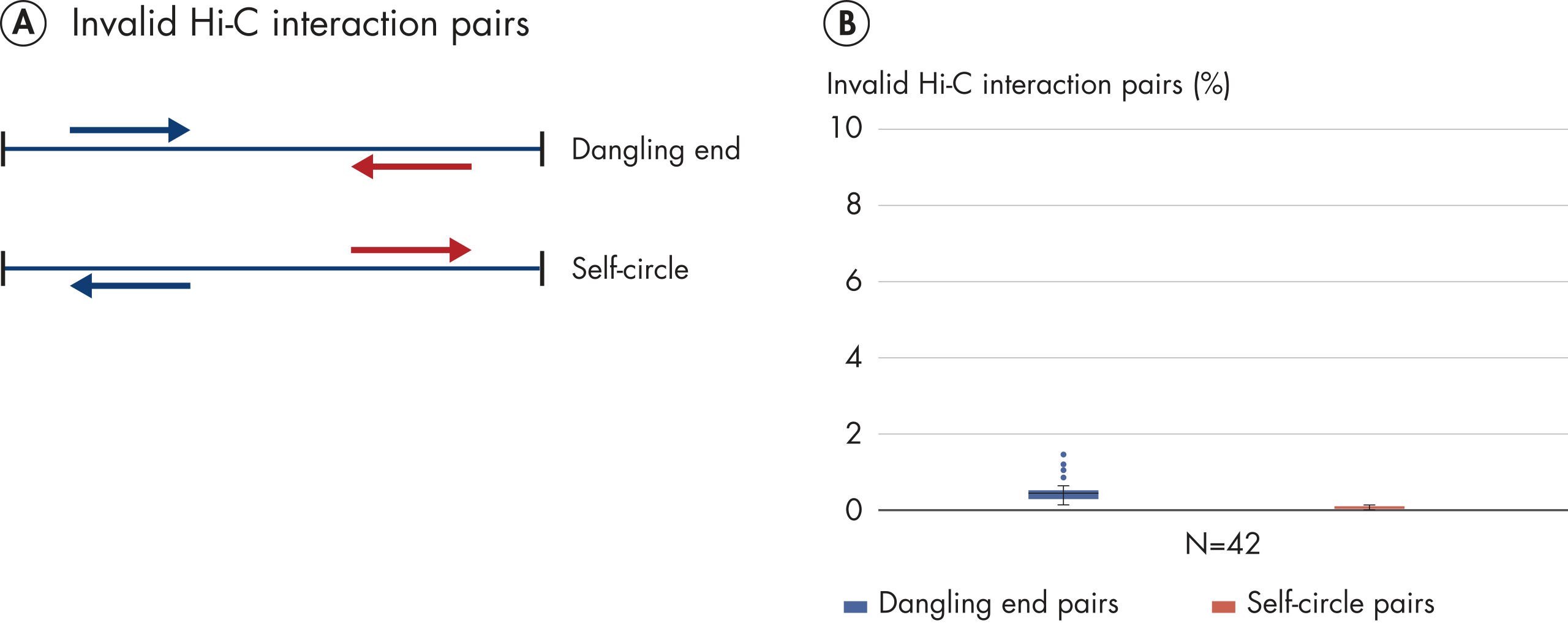
Das EpiTect Hi-C Kit generiert qualitativ hochwertige Hi-C-NGS-Bibliotheken, die sicherstellen, dass in der kostenintensiven nachgelagerten Deep-Sequencing-Analyse exzellente Daten gewonnen werden. Um die Leistung des Kits zu bewerten, wurden Sequenzierergebnisse von mehr als 40 EpiTect Hi-C-Bibliotheken analysiert. Die wichtigsten QK-Kriterien werden in den folgenden Abbildungen dargestellt: Prozentsatz der Hi-C-Ereignisse, Prozentsatz der langreichweitigen cis-Interaktionen, cis/trans-Verhältnis, Kein Bias der Strangorientierung mit dem EpiTect Hi-C Kit und Prozentsatz der gepaarten Reads, die von einem einzigen Restriktionsfragment stammen. Die Daten zeigen, dass die mit dem EpiTect Hi-C Kit erzeugten NGS-Bibliotheken im Durchschnitt weit über die Kriterien hinausgehen, die normalerweise für ein erfolgreiches Hi-C-Experiment als ausreichend gelten.
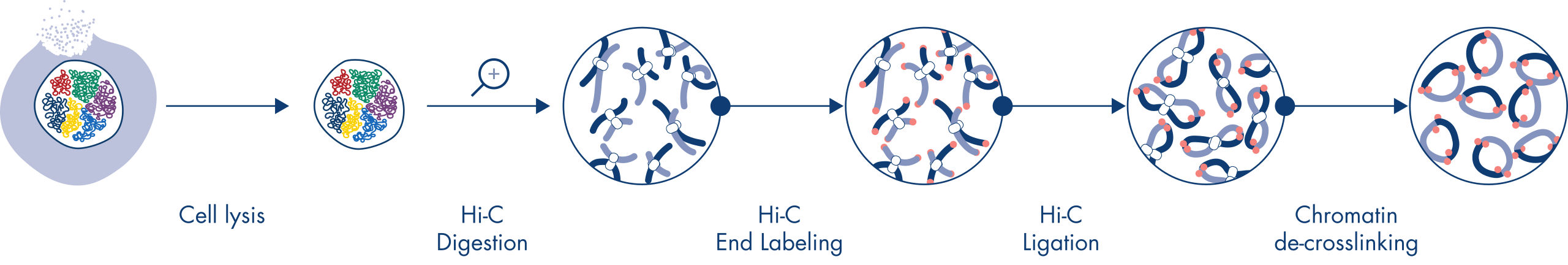
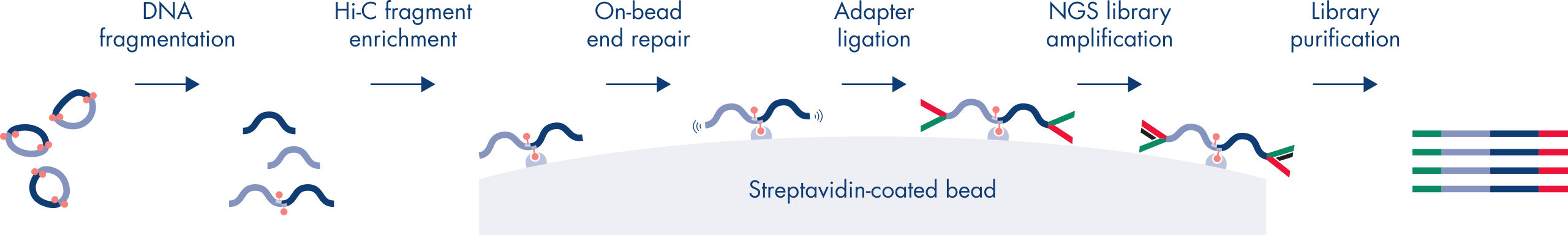
Hi-C ist ein Proximity-Ligation-Assay, der Chromatin-Interaktionen über das gesamte Genom hinweg erfasst. Das EpiTect Hi-C Kit ist eine spezielle DNA-Präparationsmethode, die die Erstellung einer Illumina-kompatiblen NGS-Bibliothek ermöglicht (siehe Abbildungen EpiTect Hi-C-Workflow – Tag 1 und EpiTect Hi-C-Workflow – Tag 2). Kurz zusammengefasst beginnt der Assay mit der Aufreinigung der Zellkerne, in denen die Chromatinkonformation durch chemische Quervernetzung von DNA-Bindungsproteinen und DNA fixiert wurde. Die DNA wird dann mit einem 4-bp-Restriktionsenzym vollständig verdaut. Offene DNA-Enden werden mit Biotin markiert und anschließend ligiert. Die Paired-End-Sequenzierung der Hi-C-Ligationsprodukte ermöglicht die Identifizierung einer sehr großen Anzahl chimärer Sequenzen aus DNA-Strängen, die räumlich eng miteinander verbunden waren. Die Wahrscheinlichkeit, dass zwei Sequenzen miteinander ligiert werden, hängt von ihrem mittleren räumlichen Abstand ab. Die Quantifizierung von Ligationsverbindungen ermöglicht die Bestimmung von DNA-Kontakthäufigkeiten, anhand derer eine hochauflösende Kartierung der Chromatinfaltung erfolgen kann.
Der EpiTect Hi-C-Workflow (siehe Abbildungen EpiTect Hi-C-Workflow – Tag 1 und EpiTect Hi-C-Workflow – Tag 2) stellt eine deutliche Verbesserung gegenüber bisher veröffentlichten Protokollen dar. Ein wochenlanges und kompliziertes Verfahren wurde in ein einfaches und robustes Protokoll umgewandelt, das innerhalb von nur 1,5 Tagen durchgeführt werden kann. Darüber hinaus wurde die erforderliche Probenmenge um eine Größenordnung reduziert, sodass Hi-C-NGS-Bibliotheken schon aus nur 5000 Zellen erstellt werden können. Das Protokoll wurde für die Bearbeitung von quervernetzten Zellen aus Säugerzellkulturen entwickelt.
Das EpiTect Hi-C-Verfahren ist eine Version der in situ (d. h. im Zellkern) durchgeführten Hi-C-Methode, bei der die Zellkerne schonend aufgereinigt und permeabilisiert werden, um den räumlichen Aufbau des Genoms während der ersten Verdau- und Ligationsschritte zu erhalten. Dieser Prozess ist von entscheidender Bedeutung, um das Hintergrundrauschen durch nicht-aussagekräftige Ligationsereignisse zu minimieren, die den Aufbau des Genoms nicht widerspiegeln. Dies liegt daran, dass intakte Zellkerne die Bewegung und zufälligen Kollisionen von quervernetzten Komplexen einschränken, sodass Ligationsereignisse vorwiegend zwischen topologisch assoziierten DNA-Fragmenten stattfinden.
Erstellung von Hi-C-NGS-Bibliotheken
Der Workflow des EpiTect Hi-C Kits besteht aus 2 Teilen, die jeweils an einem Tag abgeschlossen werden können. Die Schritte des Protokolls sind in den folgenden Tabellen zusammengefasst und in den Abbildungen EpiTect Hi-C-Workflow – Tag 1 und EpiTect Hi-C-Workflow – Tag 2 grafisch dargestellt. Die im Lieferumfang enthaltenen Illumina-Adapter haben Sequenz-Barcodes, die die Multiplex-Sequenzierung von bis zu 6 Proben ermöglichen.
Das vollständige Protokoll finden Sie in unserem ausführlichen EpiTect-Hi-C-Handbuch.
Datenanalyse
Die Hi-C-Datenanalyse wird in unserem GeneGlobe Data Analysis Center bereitgestellt. Die Ergebnisse der Hi-C-Sequenzierung können mithilfe des EpiTect Hi-C Data Analysis Portals analysiert werden, das unter Verwendung von Open-Source-Tools einen QK-Sequenzierungsreport, Hi-C-Kontaktmatrizen und eine grafische Darstellung von Chromatinkontaktkarten bereitstellt. Weitere Informationen finden Sie in unserer Bedienungsanleitung für das EpiTect Hi-C Data Analysis Portal.
Chromatinkonformation
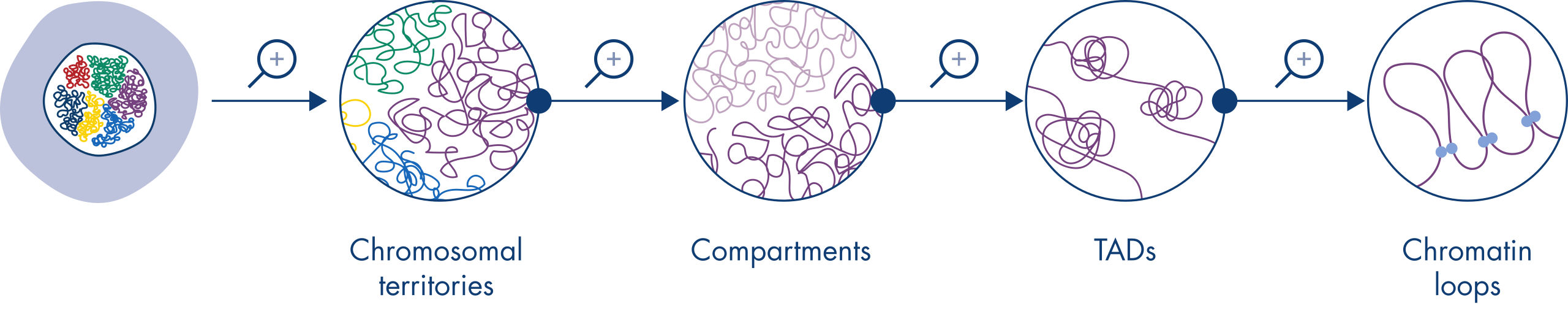
Hi-C hat sich in kürzester Zeit zu einem sehr wichtigen Instrument für die Analyse der nukleären Organisation entwickelt. Die Analyse von Hi-C-Daten hat die erstaunliche Komplexität der Genomarchitektur offengelegt, mit mehreren Ebenen der räumlichen Organisation, die das Genom mit zunehmender Auflösung in Chromosomenterritorien, chromosomale Subkompartimente, topologisch assoziierte Domänen (TADs) und DNA-Loops aufteilen (siehe Abbildung Ebenen des Chromatinaufbaus). Darüber hinaus ist die Genomorganisation dynamisch und verändert sich im Laufe der Entwicklung. Bei jedem Zelltyp sind die Chromosomen unterschiedlich gefaltet.
Chromosomale Rearrangements und Kopienzahlvarianten
Die individuellen Chromosomen sind physisch in diskrete Territorien aufgeteilt. Daher finden die von Hi-C erfassten DNA-Interaktionen in erster Linie zwischen der DNA desselben Chromosoms (in cis) und nur in geringem Maße zwischen den Chromosomen (in trans) statt. Aufgrund dieses Phänomens kann Hi-C als Assay für das gesamte Genom verwendet werden, um Translokationen und andere strukturelle Varianten von Interesse zu identifizieren. Im Vergleich zu anderen NGS-Techniken erfordert Hi-C eine extrem niedrige Abdeckung, wodurch Kosten reduziert werden können. Außerdem können Rearrangements, die schlecht kartierbare Regionen betreffen, mit Hi-C besser detektiert werden als mit NGS-Standardmethoden. Praktisch ist, dass dieselben Hi-C-Daten auch zum Nachweis von Kopienzahländerungen verwendet werden können.
Genomassemblierung – Haplotyp-Phasing
Bei der Sequenzierung und Assemblierung der Genome neuer Spezies wird die Erstellung von Sequenz-Scaffolds oft durch große Abschnitte mit repetitiven Sequenzen, die über die Reichweite der Sequenzierung hinausgehen, eingeschränkt. In Hi-C-Daten findet die überwiegende Mehrheit der Interaktionen in cis zwischen Loci auf demselben Chromosom statt. Außerdem findet ein signifikanter Teil dieser cis-Interaktionen über große Entfernungen statt, zwischen Loci, die durch Millionen von DNA-Basen voneinander getrennt sind. Diese Eigenschaften von Chromatin-Interaktionen können genutzt werden, um Sequenz-Scaffolds zu ordnen, zu orientieren und zu nahezu vollständigen Chromosomen zusammenzufügen, ohne dass dafür ein Referenzgenom erforderlich ist. Nach dem gleichen Prinzip können Hi-C-Interaktionskarten zur Erstellung diploider Genome verwendet werden, indem genetische Varianten den väterlichen und mütterlichen Schwesterchromosomen zugeordnet werden (siehe Abbildung Nachgelagerte Anwendungen von Hi-C-Sequenzierungsdaten).